Don Gass, MD, first described acute posterior multifocal placoid pigment epitheliopathy (APMPPE or AMPPE) in 1968. The clinical presentation is usually characterized by multiple, bilateral, cream-colored, placoid lesions at the level of the outer retina/retinal pigment epithelium (RPE). It is caused by ischemic changes occurring within the choriocapillaris.
Epidemiology and Etiology
 It predominantly occurs in young individuals that are in the second to fourth decades of life.
It predominantly occurs in young individuals that are in the second to fourth decades of life.
 It affects men and women equally.
It affects men and women equally.
 APMPPE is usually bilateral, but it can be quite asymmetric and the second eye may not become involved for several weeks after the first eye.
APMPPE is usually bilateral, but it can be quite asymmetric and the second eye may not become involved for several weeks after the first eye.
 APMPPE is idiopathic, but it has been associated with mumps, secondary syphilis, Lyme disease, streptococcal group A infection and anti–hepatitis B virus vaccination.
APMPPE is idiopathic, but it has been associated with mumps, secondary syphilis, Lyme disease, streptococcal group A infection and anti–hepatitis B virus vaccination.
 The disease has also been called acute multifocal ischemic choroidopathy.
The disease has also been called acute multifocal ischemic choroidopathy.
Symptoms
 Patients complain of visual disturbances, blurred vision, photopsias and scotomas.
Patients complain of visual disturbances, blurred vision, photopsias and scotomas.
 The degree of visual impairment can be variable depending upon the location of the lesions.
The degree of visual impairment can be variable depending upon the location of the lesions.
 Visual loss is due to macular involvement, and there is usually a gradual improvement in vision over the course of a few weeks.
Visual loss is due to macular involvement, and there is usually a gradual improvement in vision over the course of a few weeks.
Signs
 Systemic signs include a flu-like syndrome and headache.
Systemic signs include a flu-like syndrome and headache.
 Episcleritis, scleritis, anterior uveitis, vitreous haze, papillitis, and retinal vasculitis can occur.
Episcleritis, scleritis, anterior uveitis, vitreous haze, papillitis, and retinal vasculitis can occur.
 On fundus examination during the acute phase, the lesions are characterized by multiple yellow-white deep plaques, ranging from one-half to one optic disk diameter in size.
On fundus examination during the acute phase, the lesions are characterized by multiple yellow-white deep plaques, ranging from one-half to one optic disk diameter in size.
 Lesions appear to be at the level of the outer retina and the RPE even though the ischemic alterations involve the choriocapillaris.
Lesions appear to be at the level of the outer retina and the RPE even though the ischemic alterations involve the choriocapillaris.
 After a few days, healing starts at the center of the plaques, leaving a pigmented scar or mottled RPE.
After a few days, healing starts at the center of the plaques, leaving a pigmented scar or mottled RPE.
 New lesions can appear in the posterior pole within the first 3 weeks of disease onset.
New lesions can appear in the posterior pole within the first 3 weeks of disease onset.
 APMPPE may be associated with serous retinal detachments, mimicking Vogt-Koyanagi-Harada (VKH) disease.
APMPPE may be associated with serous retinal detachments, mimicking Vogt-Koyanagi-Harada (VKH) disease.
Differential Diagnosis
 White dot syndromes: Presumed ocular histoplasmosis syndrome, punctuate inner choroidopathy (PIC), multifocal choroiditis
White dot syndromes: Presumed ocular histoplasmosis syndrome, punctuate inner choroidopathy (PIC), multifocal choroiditis
 Sarcoidosis, syphilis, tuberculosis (TB)
Sarcoidosis, syphilis, tuberculosis (TB)
 VKH syndrome
VKH syndrome
 Sympathetic ophthalmia
Sympathetic ophthalmia
 Subretinal fibrosis/uveitis syndrome
Subretinal fibrosis/uveitis syndrome
 Serpiginous choroiditis
Serpiginous choroiditis
 Birdshot chorioretinopathy
Birdshot chorioretinopathy
Diagnostic Evaluation (Figs. 8-1 to 8-4)
 Fluorescein angiography
Fluorescein angiography
 Acute stage
Acute stage
![]() Early and intermediate hypofluorescence followed by staining and pooling in the late frames
Early and intermediate hypofluorescence followed by staining and pooling in the late frames
![]() Delay in choriocapillaris circulation
Delay in choriocapillaris circulation
 Late stage: Hyperfluorescence in early and late frames without leakage (window defect)
Late stage: Hyperfluorescence in early and late frames without leakage (window defect)
 Indocyanine green angiography (ICGA) shows hypofluorescence during the intermediate and late transit frames.
Indocyanine green angiography (ICGA) shows hypofluorescence during the intermediate and late transit frames.
 Optical coherence tomography (OCT) demonstrates nodular hyperreflectivity at the level of the photoreceptors and RPE.
Optical coherence tomography (OCT) demonstrates nodular hyperreflectivity at the level of the photoreceptors and RPE.
 Visual field testing objectively identifies the scotomata the patients report.
Visual field testing objectively identifies the scotomata the patients report.
 Interestingly, the electroretinogram (ERG) and electrooculogram (EOG) are normal.
Interestingly, the electroretinogram (ERG) and electrooculogram (EOG) are normal.
Treatment
 Usually no treatment is required.
Usually no treatment is required.
 In patients with macular involvement, systemic corticosteroids can be initiated to hasten visual recovery and immunosuppressive agents are rarely required.
In patients with macular involvement, systemic corticosteroids can be initiated to hasten visual recovery and immunosuppressive agents are rarely required.
Prognosis
 The prognosis is good with a visual acuity of 20/25 after 6 months although small scotomas may persist.
The prognosis is good with a visual acuity of 20/25 after 6 months although small scotomas may persist.
 Recurrences or exacerbations are rare.
Recurrences or exacerbations are rare.
 Macular localization may be more aggressive.
Macular localization may be more aggressive.
 Subretinal neovascularization remains a rare complication.
Subretinal neovascularization remains a rare complication.
REFERENCES
Gass JD. Acute posterior multifocal placoid pigment epitheliopathy. Arch Ophthalmol. 1968;80:177–185.
Jones BE, Jampol LM, Yannuzzi LA, et al. Relentless placoid chorioretinitis: a new entity or an unusual variant of serpiginous chorioretinitis? Arch Ophthalmol. 2000;118:931–938.
Senanayake SN, Selvadurai S, Hawkins CA, et al. Acute posterior multifocal placoid pigment epitheliopathy associated with erythema nodosum and a flu-like illness. Singapore Med J. 2008;49:e333–335.
Souka AA, Hillenkamp J, Gora F, et al. Correlation between optical coherence tomography and autofluorescence in acute posterior multifocal placoid pigment epitheliopathy. Graefes Arch Clin Exp Ophthalmol. 2006;244:1219–1223.
Figure 8-1. Color (A) and red-free (B) fundus photographs show multiple, deep yellow-white placoid lesions.
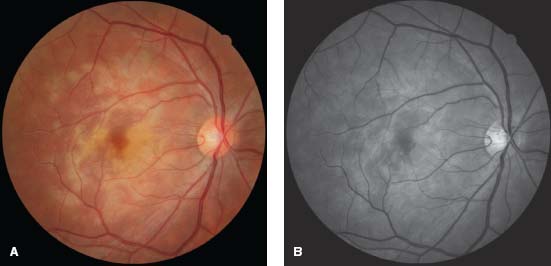
Figure 8-2. Fluorescein angiography (FA). The early frame shows hypofluorescence (A), and the late frame shows staining and pooling (B).
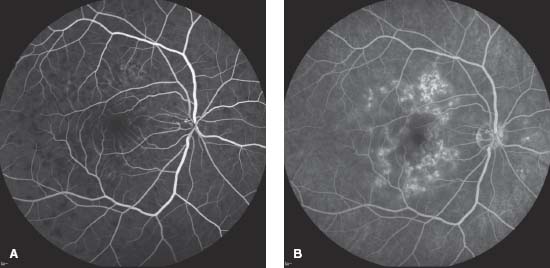
Figure 8-3. ICG demonstrates hypofluorescence in both the intermediate (A) and late (B) frames.
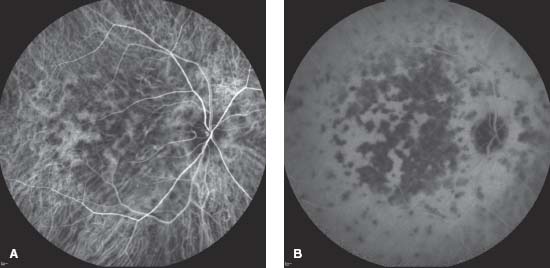
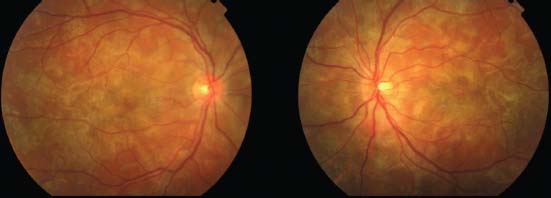
Figure 8-4. Color fundus photographs in a case of APMPPE with bilateral involvement.
SERPIGINOUS CHORIORETINOPATHY
Céline Terrada and Bahram Bodaghi
Serpiginous choroidopathy, also known as serpiginous choroiditis, geographic choroidopathy or geographic helicoid peripapillary choroidopathy, is an inflammatory disorder involving the choroid, the choriocapillaris and the RPE. It gets its name due to the serpentine pattern it develops as it progresses from the optic disk.
Epidemiology and Etiology
 Serpiginous choroiditis is a rare cause of posterior uveitis (<5% of cases in uveitis clinics), and usually occurs in the fourth to seventh decades of life.
Serpiginous choroiditis is a rare cause of posterior uveitis (<5% of cases in uveitis clinics), and usually occurs in the fourth to seventh decades of life.
 The disease is a chronic, progressive, recurrent inflammatory or infectious condition.
The disease is a chronic, progressive, recurrent inflammatory or infectious condition.
 Most patients present with unilateral vision loss, but on examination usually have bilateral disease.
Most patients present with unilateral vision loss, but on examination usually have bilateral disease.
 Males are affected slightly more often.
Males are affected slightly more often.
 The geographic distribution is worldwide.
The geographic distribution is worldwide.
 The etiology is unknown, but autoimmune and infectious hypothesis have been proposed (herpes viruses and TB).
The etiology is unknown, but autoimmune and infectious hypothesis have been proposed (herpes viruses and TB).
 There is no human leukocyte antigen (HLA) association.
There is no human leukocyte antigen (HLA) association.
Symptoms
 Patients without macular alterations and choroidal neovascularization (CNV) present with photopsias and decreased vision.
Patients without macular alterations and choroidal neovascularization (CNV) present with photopsias and decreased vision.
 Patients with macular involvement or CNV are usually symptomatic and may have metamorphopsia, scotoma and sudden vision loss.
Patients with macular involvement or CNV are usually symptomatic and may have metamorphopsia, scotoma and sudden vision loss.
Signs (Figs. 8-5 to 8-7)
 The main finding is a peripapillary choroiditis that has a characteristic serpentine appearance. In some cases, it involves the macula while sparing the peripapillary region.
The main finding is a peripapillary choroiditis that has a characteristic serpentine appearance. In some cases, it involves the macula while sparing the peripapillary region.
 The lesions are typically not multifocal as opposed to ampiginous or tuberculous serpiginous-like choroiditis.
The lesions are typically not multifocal as opposed to ampiginous or tuberculous serpiginous-like choroiditis.
 The areas of active choroiditis appear gray or white-yellow. They often appear to elevate the overlying retina.
The areas of active choroiditis appear gray or white-yellow. They often appear to elevate the overlying retina.
 The active lesions spontaneously resolve after 6 to 8 weeks and result in atrophy of the RPE and choriocapillaris. Inactive lesions appear as pigmented and atrophic scars that are usually connected to the optic disk.
The active lesions spontaneously resolve after 6 to 8 weeks and result in atrophy of the RPE and choriocapillaris. Inactive lesions appear as pigmented and atrophic scars that are usually connected to the optic disk.
 Patients may have recurrences months to years after the initial lesions and the reactivation occurs at the edges of the previous lesions.
Patients may have recurrences months to years after the initial lesions and the reactivation occurs at the edges of the previous lesions.
 One-third of affected individuals have mild inflammatory cells in the posterior vitreous. Rarely is inflammation seen in the anterior segment.
One-third of affected individuals have mild inflammatory cells in the posterior vitreous. Rarely is inflammation seen in the anterior segment.
 CNV may occur in up to 35% of cases.
CNV may occur in up to 35% of cases.
 Occasionally patients may have RPE and serous retinal detachments.
Occasionally patients may have RPE and serous retinal detachments.
Differential Diagnosis
 White dot syndromes: APMPPE or ampiginous choroiditis (which have a multifocal presentation of lesions)
White dot syndromes: APMPPE or ampiginous choroiditis (which have a multifocal presentation of lesions)
 Tuberculous serpiginous-like choroiditis (TB-SLC): Patients with TB-SLC come from areas in which TB is endemic, have significant vitritis, and usually have multifocal lesions in the periphery and posterior pole. Patients with serpiginous choroiditis have little to no vitritis, have bilateral involvement, and have a larger, more confluent lesion, which extends mainly from the disk and usually is confined to the posterior pole.
Tuberculous serpiginous-like choroiditis (TB-SLC): Patients with TB-SLC come from areas in which TB is endemic, have significant vitritis, and usually have multifocal lesions in the periphery and posterior pole. Patients with serpiginous choroiditis have little to no vitritis, have bilateral involvement, and have a larger, more confluent lesion, which extends mainly from the disk and usually is confined to the posterior pole.
 Sarcoidosis
Sarcoidosis
 Syphilis
Syphilis
 VKH syndrome
VKH syndrome
 Sympathetic ophthalmia
Sympathetic ophthalmia
 Subretinal fibrosis/uveitis syndrome
Subretinal fibrosis/uveitis syndrome
Diagnostic Evaluation
 Fundus photography is helpful to follow disease progression.
Fundus photography is helpful to follow disease progression.
 Fluorescein angiography (FA)
Fluorescein angiography (FA)
 If no CNV is present:
If no CNV is present:
![]() Areas of acute choroiditis demonstrate early hypofluorescence with blockage and diffuse late staining and leakage of dye.
Areas of acute choroiditis demonstrate early hypofluorescence with blockage and diffuse late staining and leakage of dye.
![]() Inactive, atrophic lesions appear as a window effect with a late hyperfluorescent border. Disappearance of the hyperfluorescent border suggests a recurrence.
Inactive, atrophic lesions appear as a window effect with a late hyperfluorescent border. Disappearance of the hyperfluorescent border suggests a recurrence.
 CNV in these cases shows the typical signs of a classic CNV: well-defined early hyperfluorescence with leakage in the late phase.
CNV in these cases shows the typical signs of a classic CNV: well-defined early hyperfluorescence with leakage in the late phase.
 ICGA
ICGA
 Hypoperfusion of the choroid is more extensive than shown by FA. It appears as a primary inflammatory choriocapillaropathy.
Hypoperfusion of the choroid is more extensive than shown by FA. It appears as a primary inflammatory choriocapillaropathy.
 Lesions not seen on the FA can be apparent on ICGA.
Lesions not seen on the FA can be apparent on ICGA.
 OCT can identify:
OCT can identify:
 Macular edema in rare cases
Macular edema in rare cases
 Areas of retinal atrophy (which appears to be secondary to atrophy of the RPE and choriocapillaris)
Areas of retinal atrophy (which appears to be secondary to atrophy of the RPE and choriocapillaris)
 Visual fields can be useful to identify scotomas, which can be absolute and/or relative. The visual field deficit can vary over time.
Visual fields can be useful to identify scotomas, which can be absolute and/or relative. The visual field deficit can vary over time.
 It is important to exclude TB with a tuberculin skin test (PPD), chest radiograph, and/or interferon-gamma releasing assays (e.g., QuantiFERON), especially before any immunosuppressive therapy is instituted.
It is important to exclude TB with a tuberculin skin test (PPD), chest radiograph, and/or interferon-gamma releasing assays (e.g., QuantiFERON), especially before any immunosuppressive therapy is instituted.
Treatment
 Patients with an active inflammatory component benefit from treatment.
Patients with an active inflammatory component benefit from treatment.
 Corticosteroids: High-dose systemic steroids, as well as periocular steroids, can be used to control acute inflammation.
Corticosteroids: High-dose systemic steroids, as well as periocular steroids, can be used to control acute inflammation.
 Immunomodulatory or immunosuppressive agents
Immunomodulatory or immunosuppressive agents
![]() May be considered in refractory cases or for individuals intolerant to corticosteroids
May be considered in refractory cases or for individuals intolerant to corticosteroids
![]() Mycophenolate mofetil, azathioprine, cyclosporine A and cyclophosphamide have been used in severe cases, and some investigators have recommended triple therapy with azathioprine, cyclosporine and oral prednisone.
Mycophenolate mofetil, azathioprine, cyclosporine A and cyclophosphamide have been used in severe cases, and some investigators have recommended triple therapy with azathioprine, cyclosporine and oral prednisone.
![]() Treatment must be adapted to the severity of each situation and must take into account the potential side effects.
Treatment must be adapted to the severity of each situation and must take into account the potential side effects.
 Active disease with CNV: Corticosteroids and immunosuppressive agents are usually used along with other adjunctive measures.
Active disease with CNV: Corticosteroids and immunosuppressive agents are usually used along with other adjunctive measures.
 Extrafoveal: Focal laser
Extrafoveal: Focal laser
 Juxtafoveal: Anti–vascular endothelial growth factor (VEGF) therapy, photodynamic therapy, focal laser
Juxtafoveal: Anti–vascular endothelial growth factor (VEGF) therapy, photodynamic therapy, focal laser
 Subfoveal: Anti-VEGF therapy, photodynamic therapy
Subfoveal: Anti-VEGF therapy, photodynamic therapy
 Peripapillary: Focal laser for small lesions, anti-VEGF therapy
Peripapillary: Focal laser for small lesions, anti-VEGF therapy
Prognosis
 The prognosis depends on macular involvement and complications including CNV formation.
The prognosis depends on macular involvement and complications including CNV formation.
 Unilateral or bilateral severe visual loss may be observed in 25% of cases.
Unilateral or bilateral severe visual loss may be observed in 25% of cases.
REFERENCES
Akpek EK, Jabs DA, Tessler HH, et al. Successful treatment of serpiginous choroiditis with alkylating agents. Ophthalmology. 2002;109:1506–1513.
Cardillo-Piccolino F, Grosso A, Savini E. Fundus auto-fluorescence in serpiginous choroiditis. Graefes Arch Clin Exp Ophthalmol. 2009;247:179–185.
Gupta V, Gupta A, Rao NA. Intraocular tuberculosis: an update. Surv Ophthalmol. 2007;52:561–587.
Lim WK, Buggage RR, Nussenblatt RB. Serpiginous choroiditis. Surv Ophthalmol. 2005;50:231–244.
Song MH, Roh YJ. Intravitreal ranibizumab for choroidal neovascularization in serpiginous choroiditis. Eye. 2008.
Vasconcelos-Santos DV, Rao PK, Davies JB, et al. Clinical features of tuberculous serpiginouslike choroiditis in contrast to classic serpiginous choroiditis. Arch Ophthalmol. 2010;128:853–858.
Figure 8-5. Color fundus photograph (A) and red-free photo (B) show the sequelae of choroiditis. Note the geographic pigmented and atrophic scars connected to the optic disk. C. The early frame of the fluorescein angiogram shows hypofluorescence likely due to limited perfusion. D. The late frame demonstrates staining and with a hyperfluorescent border, characteristic of inactive serpiginous.
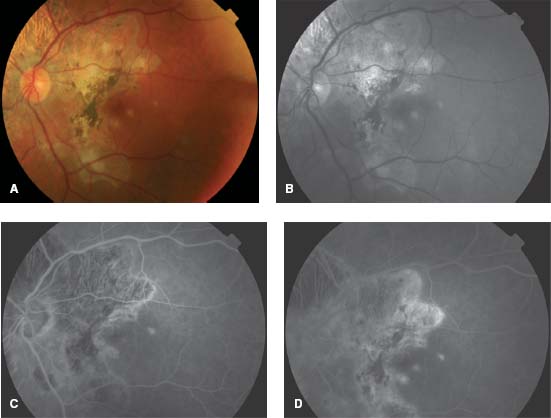
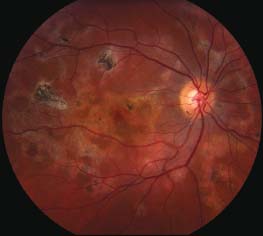
Figure 8-6. This patient with a previous diagnosis of serpiginous chorioretinopathy came in with further vision changes. A. The color photograph reveals darker areas consistent with earlier scarring, as well as yellow areas of active chorioretinitis. B. The early frame of the fluorescein angiogram reveals RPE window defects in the area of previous scarring. The new lesions block early. C. The new lesions stain in the late frames of the angiogram. (Courtesy of MidAtlantic Retina, the Retina Service of Wills Eye Institute.)
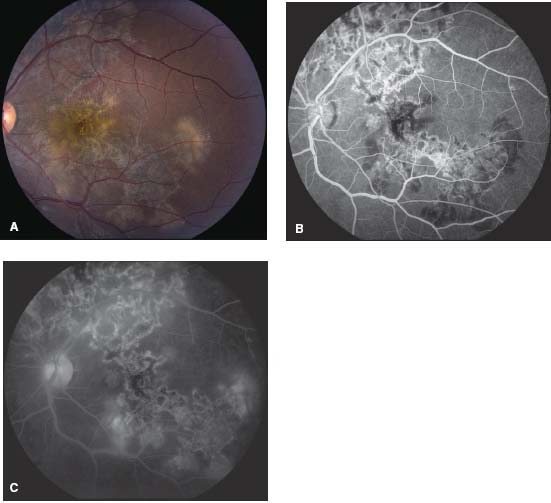
Figure 8-7. A 27-year-old woman had serpiginous choroiditis that progressed despite numerous immunosuppressive agents. Her vision was 20/400 in this eye. (Courtesy of Sunir Garg, MD.)
MULTIPLE EVANESCENT WHITE DOT SYNDROME
Céline Terrada and Bahram Bodaghi
Multiple evanescent white dot syndrome (MEWDS) is a rare, acute, multifocal inflammatory retinochoroidopathy. It spontaneously resolves with an excellent visual outcome.
Epidemiology and Etiology
 It occurs mainly in young, healthy young women in the second to fourth decades of life.
It occurs mainly in young, healthy young women in the second to fourth decades of life.
 The etiology remains unknown, however an infectious etiology has been suspected, as one-third of patients have a viral prodrome.
The etiology remains unknown, however an infectious etiology has been suspected, as one-third of patients have a viral prodrome.
 Patients often have mild myopia.
Patients often have mild myopia.
Symptoms
 Photopsias, often in the temporal visual field
Photopsias, often in the temporal visual field
 Blurry vision, with visual acuity ranging from 20/20 to 20/400
Blurry vision, with visual acuity ranging from 20/20 to 20/400
 Central microscotoma
Central microscotoma
Signs (Figs. 8-8 to 8-10)
 Multiple small dots in the fundus at the level of the outer retina and the RPE
Multiple small dots in the fundus at the level of the outer retina and the RPE
 An orange granularity of the fovea
An orange granularity of the fovea
 No anterior segment inflammation, but some patients have a mild vitritis
No anterior segment inflammation, but some patients have a mild vitritis
 A mild afferent pupillary defect may occur
A mild afferent pupillary defect may occur
 Optic disk edema
Optic disk edema
 Usually monophasic, unilateral and self-limited
Usually monophasic, unilateral and self-limited
Differential Diagnosis
 Inflammatory white dot syndromes
Inflammatory white dot syndromes
 Acute idiopathic blind spot enlargement syndrome (AIBSE)
Acute idiopathic blind spot enlargement syndrome (AIBSE)
 Acute zonal occult outer retinopathy (AZOOR)
Acute zonal occult outer retinopathy (AZOOR)
Diagnostic Evaluation
 White dots
White dots
 FA demonstrates both early and late hyperfluorescence of the dots and late staining of the optic disk
FA demonstrates both early and late hyperfluorescence of the dots and late staining of the optic disk
 ICG
ICG
![]() Intermediate/late hypofluorescent spots scattered throughout the posterior pole and mid-periphery.
Intermediate/late hypofluorescent spots scattered throughout the posterior pole and mid-periphery.
![]() The lesions are more numerous on ICGA than seen clinically or on FA.
The lesions are more numerous on ICGA than seen clinically or on FA.
![]() Intermediate transient pinpoint hyperfluorescence.
Intermediate transient pinpoint hyperfluorescence.
 Fundus autofluorescence
Fundus autofluorescence
![]() In the acute phase of the disease, there are small areas of hypoautofluorescence around the disk and posterior pole and hyperfluorescent areas corresponding to the white dots. These spots evolve over time and may persist or fade.
In the acute phase of the disease, there are small areas of hypoautofluorescence around the disk and posterior pole and hyperfluorescent areas corresponding to the white dots. These spots evolve over time and may persist or fade.
![]() The lesions are more numerous than clinically seen.
The lesions are more numerous than clinically seen.
 OCT demonstrates defects located at the level of the outer retina and RPE. The areas corresponding to the white spots show interruption or signal attenuation of the photoreceptor inner segment/outer segment band.
OCT demonstrates defects located at the level of the outer retina and RPE. The areas corresponding to the white spots show interruption or signal attenuation of the photoreceptor inner segment/outer segment band.
 Granularity of the macula
Granularity of the macula
 FA shows hyperfluorescence throughout the angiogram.
FA shows hyperfluorescence throughout the angiogram.
 ICGA demonstrates intermediate/late hypofluorescence.
ICGA demonstrates intermediate/late hypofluorescence.
 Fundus autofluorescence shows pinpoint areas of increased or decreased autofluorescence.
Fundus autofluorescence shows pinpoint areas of increased or decreased autofluorescence.
Treatment
 Observation as MEWDS spontaneously resolves over a period of several weeks to months with return of normal vision in nearly all patients.
Observation as MEWDS spontaneously resolves over a period of several weeks to months with return of normal vision in nearly all patients.
Prognosis
 Very good.
Very good.
 Mild pigmentary changes may be seen after recovery, and some patients will have persistent visual field or color deficits.
Mild pigmentary changes may be seen after recovery, and some patients will have persistent visual field or color deficits.
 Submacular CNV may rarely develop.
Submacular CNV may rarely develop.
REFERENCES
Dell’Omo R, Mantovani A, Wong R, et al. Natural evolution of fundus autofluorescence findings in multiple evanescent white dot syndrome. Retina. 2010;30:1479–1487.
Gass JD, Hamed LM. Acute macular neuroretinopathy and multiple evanescent white dot syndrome occurring in the same patient. Arch Ophthalmol. 1989;107:189–193.
Jampol LM, Sieving PA, Pugh D, et al. Multiple evanescent white dot syndrome. I. Clinical findings. Arch Ophthalmol. 1984;102:671–674.
Mamalis N, Daily MJ. Multiple evanescent white dot syndrome. A report of eight cases. Ophthalmology. 1987;94:1209–1212.
Nguyen MH, Witkin AJ, Reichel E, et al. Microstructural abnormalities in MEWDS demonstrated by ultrahigh resolution optical coherence tomography. Retina. 2007;27:414–418.
Figure 8-8. Color photograph showing numerous small, yellow-white spots scattered throughout the posterior pole in a patient with MEWDS.
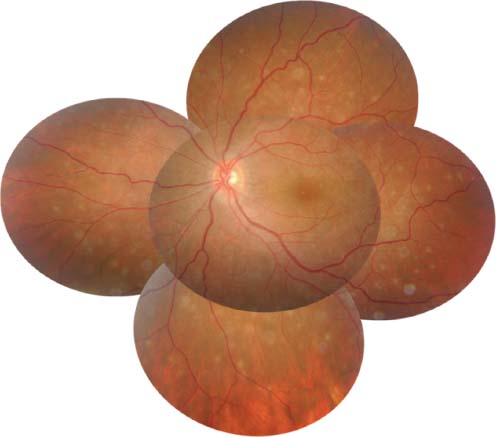
Figure 8-9. FA early (A) and late (B) frames showing hyperfluorescence and disk staining. C. Late frame ICGA, showing numerous hypofluorescent spots. Note there are more spots seen on ICGA than on FA.
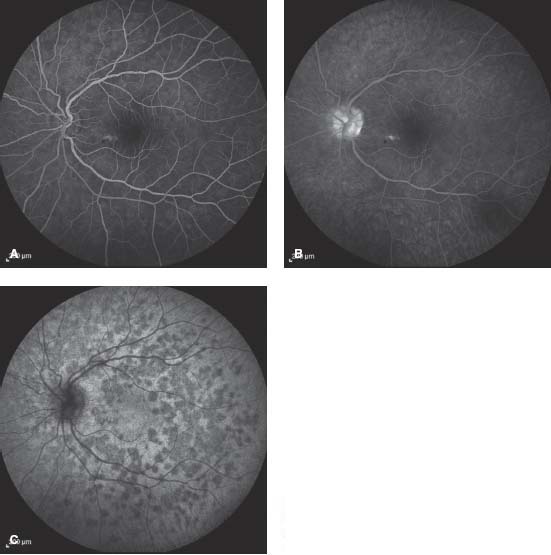
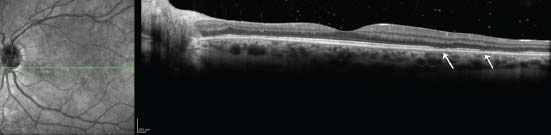
Figure 8-10. SD-OCT in MEWDS patient illustrating interruption or signal attenuation of the photoreceptor inner segment/outer segment band (arrows).
MULTIFOCAL CHOROIDITIS/SUBRETINAL FIBROSIS SYNDROME
Céline Terrada and Bahram Bodaghi
Multifocal choroiditis and panuveitis syndrome (MFCP) was described by Dreyer and Gass in 1984. It is a rare, idiopathic, inflammatory choroidal disease, mimicking presumed ocular histoplasmosis syndrome (OHS), which affects otherwise healthy young adults. MFCP, PIC, MEWDS and AIBES may constitute different clinical presentations of the same disease but this hypothesis has not been validated.
Epidemiology and Etiology
 The disease usually affects Caucasian adults (80% of cases).
The disease usually affects Caucasian adults (80% of cases).
 MFCP usually occurs in the third to fourth decades of life, and affects women more than men (approximately 3:1), and most patients have some degree of myopia.
MFCP usually occurs in the third to fourth decades of life, and affects women more than men (approximately 3:1), and most patients have some degree of myopia.
 The disease is bilateral in approximately 80% of cases, but it can be asymmetric or unilateral at presentation.
The disease is bilateral in approximately 80% of cases, but it can be asymmetric or unilateral at presentation.
 It occurs worldwide.
It occurs worldwide.
 There are usually no other systemic findings.
There are usually no other systemic findings.
 There is no well-defined HLA-association. It may be virally induced.
There is no well-defined HLA-association. It may be virally induced.
Symptoms
 Patients describe photopsias, floaters, and/or an enlarged blind spot; rarely some patients may remain asymptomatic.
Patients describe photopsias, floaters, and/or an enlarged blind spot; rarely some patients may remain asymptomatic.
 With macular involvement or submacular choroidal neovascular membrane formation, patients describe metamorphopsia, subjective scotomas and/or sudden central vision loss.
With macular involvement or submacular choroidal neovascular membrane formation, patients describe metamorphopsia, subjective scotomas and/or sudden central vision loss.
Signs
 Anterior chamber flare and cells are variable in intensity but are usually mild.
Anterior chamber flare and cells are variable in intensity but are usually mild.
 Vitreous haze is common and is useful to distinguish MFC from ocular histoplasmosis.
Vitreous haze is common and is useful to distinguish MFC from ocular histoplasmosis.
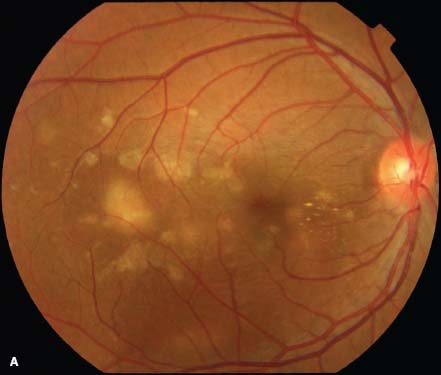
 Multiple choroidal lesions (20 to 100) are randomly distributed throughout the posterior pole and mid-periphery. Active lesions have a “creamy” appearance, while older lesions appear atrophic and can become pigmented at their edges. The lesions appear to be at the level of the RPE and choroid.
Multiple choroidal lesions (20 to 100) are randomly distributed throughout the posterior pole and mid-periphery. Active lesions have a “creamy” appearance, while older lesions appear atrophic and can become pigmented at their edges. The lesions appear to be at the level of the RPE and choroid.
 The lesion size ranges from 50 to 350 μm in diameter. They can enlarge and become coalescent.
The lesion size ranges from 50 to 350 μm in diameter. They can enlarge and become coalescent.
 CNV is present in 30% of cases. CNV is characterized by the presence of subretinal fluid, subretinal hemorrhage and subretinal exudate.
CNV is present in 30% of cases. CNV is characterized by the presence of subretinal fluid, subretinal hemorrhage and subretinal exudate.
 Cystoid macular edema (CME), epiretinal membrane (ERM), optic disk edema, enlarged blind spots and peripapillary scarring also occur.
Cystoid macular edema (CME), epiretinal membrane (ERM), optic disk edema, enlarged blind spots and peripapillary scarring also occur.
 Subretinal fibrosis may link the atrophic scars and is probably due to subclinical progression of the disease.
Subretinal fibrosis may link the atrophic scars and is probably due to subclinical progression of the disease.
Differential Diagnosis
 White dot syndromes
White dot syndromes
 Ocular histoplasmosis: Unlike MFC, OHS does not have anterior uveitis or vitritis.
Ocular histoplasmosis: Unlike MFC, OHS does not have anterior uveitis or vitritis.
 Punctate inner choroidopathy: Many feel that this is a variant of MFC. In PIC, the lesions tend to be deeper and more punched out, and these patients have less vitritis.
Punctate inner choroidopathy: Many feel that this is a variant of MFC. In PIC, the lesions tend to be deeper and more punched out, and these patients have less vitritis.
 Birdshot retinochoroidopathy: These patients are usually HLA-A29 positive, are older, and have larger and less discrete lesions.
Birdshot retinochoroidopathy: These patients are usually HLA-A29 positive, are older, and have larger and less discrete lesions.
 Sarcoidosis, syphilis, TB
Sarcoidosis, syphilis, TB
 VKH syndrome
VKH syndrome
 Sympathetic ophthalmia
Sympathetic ophthalmia
 Subretinal fibrosis/uveitis syndrome
Subretinal fibrosis/uveitis syndrome
 Serpiginous choroiditis
Serpiginous choroiditis
Diagnostic Evaluation (Figs. 8-11 to 8-13)
 FA
FA
 Acute choroiditis appears as early hypofluorescence with staining in late frames.
Acute choroiditis appears as early hypofluorescence with staining in late frames.
 Late (chronic) choroiditis has early hyperfluorescence that persists in the late frames (window effect).
Late (chronic) choroiditis has early hyperfluorescence that persists in the late frames (window effect).
 Staining of the optic disk and vessels
Staining of the optic disk and vessels
 CME
CME
 If a CNV is present, there is well-defined early hyperfluorescence with late leakage.
If a CNV is present, there is well-defined early hyperfluorescence with late leakage.
 ICGA
ICGA
 Large and small hypofluorescent inflammatory lesions that may cluster around the disk
Large and small hypofluorescent inflammatory lesions that may cluster around the disk
 There are more lesions on ICGA than seen clinically.
There are more lesions on ICGA than seen clinically.
 Fundus autofluorescence shows hypoautofluorescent scars.
Fundus autofluorescence shows hypoautofluorescent scars.
 The larger lesions (>125 μm) appear as atrophic scars.
The larger lesions (>125 μm) appear as atrophic scars.
 Interestingly, there are often hundreds of smaller lesions (<125 μm) that are not clinically apparent.
Interestingly, there are often hundreds of smaller lesions (<125 μm) that are not clinically apparent.
 OCT demonstrates nodular hyperreflectivity at the level of the photoreceptors and RPE.
OCT demonstrates nodular hyperreflectivity at the level of the photoreceptors and RPE.
 Visual field testing objectively identifies the areas of visual field loss and periodic testing may help to monitor disease progression.
Visual field testing objectively identifies the areas of visual field loss and periodic testing may help to monitor disease progression.
 ERG finding are often normal or mildly depressed and are nonspecific. Multifocal ERG can show diffuse depression in addition to focal areas of greater depression, which correspond to the scotomas on the visual field.
ERG finding are often normal or mildly depressed and are nonspecific. Multifocal ERG can show diffuse depression in addition to focal areas of greater depression, which correspond to the scotomas on the visual field.
Treatment
 Uveitic component
Uveitic component
 Corticosteroids
Corticosteroids
![]() Systemic corticosteroids are effective to control acute inflammation.
Systemic corticosteroids are effective to control acute inflammation.
![]() Periocular and intravitreal injections and sustained-release devices may be useful.
Periocular and intravitreal injections and sustained-release devices may be useful.
 Immunomodulatory therapy (IMT)
Immunomodulatory therapy (IMT)
![]() Given the chronic nature of MFC, consideration should be given to IMT, especially in cases of inflammation which cannot be controlled with low-dose steroids or for patients who are intolerant to corticosteroids.
Given the chronic nature of MFC, consideration should be given to IMT, especially in cases of inflammation which cannot be controlled with low-dose steroids or for patients who are intolerant to corticosteroids.
![]() Methotrexate, mycophenolate mofetil and azathioprine have been used with success.
Methotrexate, mycophenolate mofetil and azathioprine have been used with success.
![]() IMT has been found to greatly reduce the risk of macular complications, including CME, ERM and CNV formation.
IMT has been found to greatly reduce the risk of macular complications, including CME, ERM and CNV formation.
 CNV: Corticosteroids and IMT may reduce recurrences by controlling the inflammatory state.
CNV: Corticosteroids and IMT may reduce recurrences by controlling the inflammatory state.
 Extrafoveal CVNM can be treated with focal laser.
Extrafoveal CVNM can be treated with focal laser.
 Juxta- and subfoveal CNVM have been treated with anti-VEGF agents, photodynamic therapy and intravitreal triamcinolone injection. Submacular surgery may still be useful in cases with vision worse than 20/100 that did not respond to other treatments.
Juxta- and subfoveal CNVM have been treated with anti-VEGF agents, photodynamic therapy and intravitreal triamcinolone injection. Submacular surgery may still be useful in cases with vision worse than 20/100 that did not respond to other treatments.
 Peripapillary CNV may be treated with focal laser for small lesions and anti-VEGF therapy.
Peripapillary CNV may be treated with focal laser for small lesions and anti-VEGF therapy.
 Spontaneous resolution of the active neovascularization has been reported.
Spontaneous resolution of the active neovascularization has been reported.
Prognosis
 Seventy-five percent of patients develop permanent visual loss in at least one eye due to subfoveal lesions, CNVM, chronic CME or subretinal fibrosis.
Seventy-five percent of patients develop permanent visual loss in at least one eye due to subfoveal lesions, CNVM, chronic CME or subretinal fibrosis.
 Therapy with systemic corticosteroids and IMT has improved the prognosis:
Therapy with systemic corticosteroids and IMT has improved the prognosis:
 Eighty-three percent reduction in risk of macular complications
Eighty-three percent reduction in risk of macular complications
 Ninety-two percent reduction in the risk of developing vision loss to less than 20/200 in the affected eye
Ninety-two percent reduction in the risk of developing vision loss to less than 20/200 in the affected eye
REFERENCES
Dreyer RF, Gass DJ. Multifocal choroiditis and panuveitis. A syndrome that mimics ocular histoplasmosis. Arch Ophthalmol. 1984;102:1776–1784.
Fine HF, Zhitomirshy I, Freund KB, et al. Bevacizumab (Avastin) and ranibizumab (Lucentis) for choroidal neovascularization in multifocal choroiditis. Retina. 2009;29:8–12.
Michel SS, Ekong A, Baltazis S, et al. Multifocal choroiditis and panuveitis: immunomodulatory therapy. Ophthalmology. 2002;109:378–383.
Parnell JR, Jampol LM, Yanuzzi LA, et al. Differentiation between presumed ocular histoplasmosis syndrome and multifocal choroiditis with panuveitis based on morphology and photographed fundus lesions and fluorescein angiography. Arch Ophthalmol. 2001;119:208–212.
Thorne JE, Wittenberg S, Jabs DA, et al. Multifocal choroiditis with panuveitis: incidence of ocular complications and loss of visual acuity. Ophthalmology. 2006;113:2310–2316.
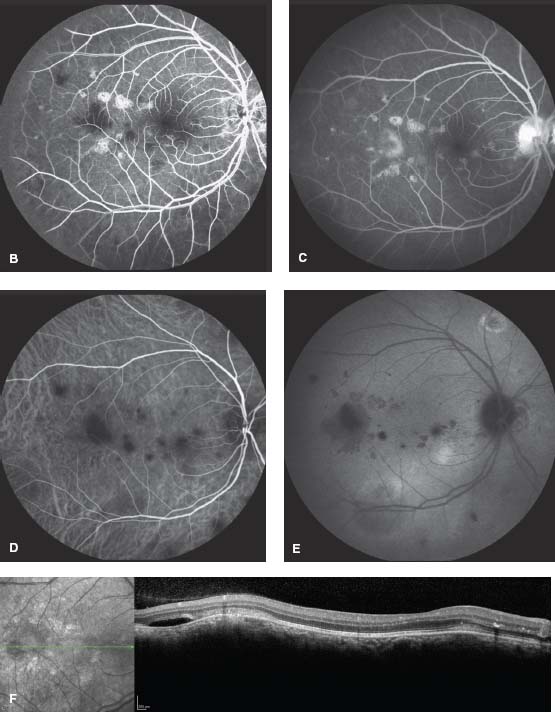
Figure 8-11. A. MFC with multiple creamy lesions are typical of subacute MFC. The fluorescein angiograms show hypofluorescence in the early frames (B) and staining in the late frames (C). ICGA shows hypocyanescent lesions both early (D) and late (E). Often more lesions are present on ICGA than on color photographs or FA. F. The OCT shows hyperreflective lesions in the outer retina/RPE. There is subretinal fluid temporal to the fovea.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


