Episcleritis is a self-limited, generally benign inflammation of the episclera.
Etiology and Epidemiology
 Episcleritis occurs most frequently in young to middle-aged women (20 to 40 years old).
Episcleritis occurs most frequently in young to middle-aged women (20 to 40 years old).
 It is frequently unilateral (70%).
It is frequently unilateral (70%).
 The etiology is unknown in most cases, but it is believed to be immune mediated, and it is occasionally associated with systemic disease.
The etiology is unknown in most cases, but it is believed to be immune mediated, and it is occasionally associated with systemic disease.
Symptoms
 Redness, minimal eye pain
Redness, minimal eye pain
 Foreign-body sensation
Foreign-body sensation
 Minimal to no changes in vision
Minimal to no changes in vision
Signs
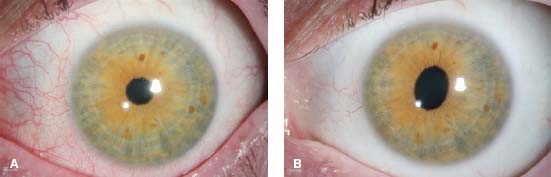
 Bright red or salmon-pink color in natural light (Fig. 3-1)
Bright red or salmon-pink color in natural light (Fig. 3-1)
 The superficial episcleral vessels are injected, and the redness can be sectoral (70%), diffuse, or, rarely, nodular.
The superficial episcleral vessels are injected, and the redness can be sectoral (70%), diffuse, or, rarely, nodular.
 The vessels are easily mobile with a cotton-tipped applicator and blanch with topical administration of 10% phenylephrine.
The vessels are easily mobile with a cotton-tipped applicator and blanch with topical administration of 10% phenylephrine.
 Small peripheral corneal opacities are present in 10% of cases.
Small peripheral corneal opacities are present in 10% of cases.
Differential Diagnosis
 Scleritis
Scleritis
 In contrast to scleritis, episcleritis has minimal pain, less common systemic disease association, and essentially no complications.
In contrast to scleritis, episcleritis has minimal pain, less common systemic disease association, and essentially no complications.
 Pinguecula
Pinguecula
 Phlyctenule
Phlyctenule
 Subconjunctival hemorrhage
Subconjunctival hemorrhage
 Conjunctival neoplasia
Conjunctival neoplasia
 Anterior uveitis
Anterior uveitis
Diagnostic Evaluation
 Although systemic disease is not commonly associated with episcleritis, a thorough review of systems and targeted systemic workup is indicated. Systemic workup can be considered in recurrent cases. Up to 30% of patients are found to have an underlying disease association.
Although systemic disease is not commonly associated with episcleritis, a thorough review of systems and targeted systemic workup is indicated. Systemic workup can be considered in recurrent cases. Up to 30% of patients are found to have an underlying disease association.
This contribution to the work was done as part of the authors’ official duties as NIH employees and is a work of the United States Government.
Treatment
 Episodes are self-limited and rarely require treatment beyond artificial tears, topical NSAIDs, and topical corticosteroids. If the episcleritis does not resolve promptly with therapy, consider another diagnosis.
Episodes are self-limited and rarely require treatment beyond artificial tears, topical NSAIDs, and topical corticosteroids. If the episcleritis does not resolve promptly with therapy, consider another diagnosis.
Prognosis
 The prognosis is usually very good.
The prognosis is usually very good.
 Episodes may recur but rarely are there lasting sequelae.
Episodes may recur but rarely are there lasting sequelae.
REFERENCES
Foster CS, and Sainz de la Maza M. Clinical consideration of episcleritis and scleritis. In The Sclera. New York: Springer-Verlag; 1994:107.
Jabs DA, Mudun A, Dunn JP, et al. Episcleritis and scleritis: clinical features and treatment results. Ophthalmology. 2000;130:469–476.
Sainz de la Maza M, Jabbur NS, Foster CS. Severity of scleritis and episcleritis. Ophthalmology. 1994;101:389–396.
Figure 3-1. Episcleritis. A. This patient has episcleritis with a salmon pink hue. B. The same eye with blanching of the episclera after administration of 10% phenylephrine.
SCLERITIS
Theresa Larson and H. Nida Sen
Scleritis is characterized by inflammation and edema of scleral and episcleral tissue. It is classified as anterior or posterior, and further subclassified as diffuse, nodular, and necrotizing (Table 3-1).
ANTERIOR SCLERITIS
Etiology and Epidemiology
 Anterior scleritis is the most common form, making up 80% to 85% of all scleritis cases. Diffuse and nodular scleritis occur with equal frequency.
Anterior scleritis is the most common form, making up 80% to 85% of all scleritis cases. Diffuse and nodular scleritis occur with equal frequency.
 Scleritis occurs most frequently in middle-aged women (40 to 60 years old)
Scleritis occurs most frequently in middle-aged women (40 to 60 years old)
 25% to 50% of patients have a history of systemic disease, most commonly rheumatoid arthritis (Table 3-2).
25% to 50% of patients have a history of systemic disease, most commonly rheumatoid arthritis (Table 3-2).
 Necrotizing scleritis is the most severe and destructive form of scleritis and is most often associated with sight-threatening sequelae.
Necrotizing scleritis is the most severe and destructive form of scleritis and is most often associated with sight-threatening sequelae.
 Necrotizing scleritis is divided into “with inflammation” and “without inflammation” (scleromalacia perforans).
Necrotizing scleritis is divided into “with inflammation” and “without inflammation” (scleromalacia perforans).
Table 3-1. Scleritis Clinical Subtypes and Their Prevalence
| Anterior scleritis | 80%–85% |
| Diffuse | 40%–45% |
| Nodular | 40% |
| Necrotizing scleritis | 10%–15% |
| With inflammation | 10% |
| Without inflammation (scleromalacia perforans) | 1%–5% |
| Posterior scleritis | 1%–5% |
Adapted from Nussenblatt RB, Whitcup SM. Uveitis: Fundamentals and Clinical Practice, 4th ed. Philadelphia: Elsevier; 2010.
This contribution to the work was done as part of the authors’ official duties as NIH employees and is a work of the United States Government.
Symptoms
 Patients present with a red, painful eye. They may have a boring eye pain.
Patients present with a red, painful eye. They may have a boring eye pain.
 Vision loss is rare.
Vision loss is rare.
 Patients with scleromalacia perforans typically have no pain.
Patients with scleromalacia perforans typically have no pain.
Signs
 The sclera has a violaceous hue in natural light with inflamed vessels that are immobile with a cotton-tipped applicator (Fig. 3-2). It is easier to appreciate scleritis by looking at the eye without the slit lamp.
The sclera has a violaceous hue in natural light with inflamed vessels that are immobile with a cotton-tipped applicator (Fig. 3-2). It is easier to appreciate scleritis by looking at the eye without the slit lamp.
 Areas of repeated attacks may demonstrate scleral thinning with a bluish hue.
Areas of repeated attacks may demonstrate scleral thinning with a bluish hue.
 Eyes with diffuse scleritis have generalized edema, while eyes with sectoral or nodular scleritis have localized erythema (Fig. 3-3).
Eyes with diffuse scleritis have generalized edema, while eyes with sectoral or nodular scleritis have localized erythema (Fig. 3-3).
 Necrotizing scleritis has a white avascular area surrounded by injection and edema (Fig. 3-4).
Necrotizing scleritis has a white avascular area surrounded by injection and edema (Fig. 3-4).
 Topical (10%) phenylephrine will not blanch the vessels in scleritis.
Topical (10%) phenylephrine will not blanch the vessels in scleritis.
Differential Diagnosis
 Episcleritis
Episcleritis
 Subconjunctival hemorrhage
Subconjunctival hemorrhage
 Conjunctival mucosa–associated lymphoid tissue lymphoma
Conjunctival mucosa–associated lymphoid tissue lymphoma
 Sentinel vessels
Sentinel vessels
 Anterior uveitis or panuveitis
Anterior uveitis or panuveitis
 Acute angle-closure glaucoma
Acute angle-closure glaucoma
 Keratitis
Keratitis
 Endophthalmitis
Endophthalmitis
 Carotid or dural sinus fistula
Carotid or dural sinus fistula
Diagnostic Evaluation
 Testing is guided by history and review of systems.
Testing is guided by history and review of systems.
 Rheumatoid factor, anti-citrullinated cyclic protein (CCP), classical antineutrophil cytoplasmic antibody (c-ANCA), protoplasmic-staining antineutrophil cytoplasmic antibodies (p-ANCA), myeloperoxidase, antinuclear antibody (ANA), and anti-dsDNA may be considered if an underlying rheumatologic disease is suspected.
Rheumatoid factor, anti-citrullinated cyclic protein (CCP), classical antineutrophil cytoplasmic antibody (c-ANCA), protoplasmic-staining antineutrophil cytoplasmic antibodies (p-ANCA), myeloperoxidase, antinuclear antibody (ANA), and anti-dsDNA may be considered if an underlying rheumatologic disease is suspected.
Table 3-2. Systemic Disease Associations
| Noninfectious | Infectious | Other |
| Connective tissue disease | Herpes zoster ophthalmicus | Gout |
| Rheumatoid arthritis Juvenile rheumatoid arthritis Reiter’s syndrome Systemic lupus erythematosus Relapsing polychondritis Polymyositis Inflammatory bowel disease Spondyloarthropathy | Herpes simplex keratitis Acanthamoeba keratitis Syphilis Lyme disease Bartonellosis Tuberculosis | Rosacea Foreign body reaction Drugs (bisphosphonates) |
| Vasculitides | ||
| Wegner’s granulomatosis Polyarteritis nodosa Allergic angiitis of Churg-Strauss Cogan syndrome Takayasu disease Adamantiades—Behçet’s disease Sarcoidosis |
 Rule out infectious causes with the appropriate tests, including: fluorescent treponemal antibody absorption (FTA-Abs), rapid plasma reagin (RPR) or Venereal Disease Research Laboratory (VDRL), purified protein derivative (PPD) and/or QuantiFERON, trauma, and foreign body.
Rule out infectious causes with the appropriate tests, including: fluorescent treponemal antibody absorption (FTA-Abs), rapid plasma reagin (RPR) or Venereal Disease Research Laboratory (VDRL), purified protein derivative (PPD) and/or QuantiFERON, trauma, and foreign body.
 Orbital CT should be considered in atypical cases.
Orbital CT should be considered in atypical cases.
Treatment
 Oral NSAIDs for milder (nonnecrotizing) scleritis cases
Oral NSAIDs for milder (nonnecrotizing) scleritis cases
 Nodular scleritis often can respond to an injection of triamcinolone over the nodule. Periocular triamcinolone can also be used selectively for cases of diffuse scleritis.
Nodular scleritis often can respond to an injection of triamcinolone over the nodule. Periocular triamcinolone can also be used selectively for cases of diffuse scleritis.
 Oral prednisone
Oral prednisone
 Immunosuppressive therapy is indicated for recurrent or severe cases (particularly for necrotizing scleritis).
Immunosuppressive therapy is indicated for recurrent or severe cases (particularly for necrotizing scleritis).
 ANCA positive patients may have a more severe course of disease and thus require more aggressive therapy.
ANCA positive patients may have a more severe course of disease and thus require more aggressive therapy.
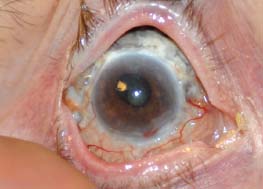
 In patients with necrotizing scleritis associated with a systemic disorder such as rheumatoid arthritis (Fig. 3-5) or Wegener’s granulomatosis, aggressive systemic therapy is necessary because it is associated with a high mortality rate without systemic treatment.
In patients with necrotizing scleritis associated with a systemic disorder such as rheumatoid arthritis (Fig. 3-5) or Wegener’s granulomatosis, aggressive systemic therapy is necessary because it is associated with a high mortality rate without systemic treatment.
Prognosis
 The prognosis varies based on the site of inflammation, associated complications, underlying conditions, and response to therapy. Diffuse anterior scleritis has the best prognosis, whereas necrotizing scleritis has the worst prognosis with the highest rates of visual loss and complications.
The prognosis varies based on the site of inflammation, associated complications, underlying conditions, and response to therapy. Diffuse anterior scleritis has the best prognosis, whereas necrotizing scleritis has the worst prognosis with the highest rates of visual loss and complications.
REFERENCES
Albini TA, Zamir E, Read RW, et al. Evaluation of subconjunctival triamcinolone for nonnecrotizing anterior scleritis. Ophthalmology. 2005;112(10):1814–1820.
Foster CS, Sainz de la Maza M. Clinical consideration of episcleritis and scleritis. In The Sclera. New York: Springer-Verlag; 1994:107.
Foster CS, Forstot SL, Wilson LA. Mortality rate in rheumatoid arthritis patients developing necrotizing scleritis or peripheral ulcerative keratitis: effects of immunosuppression. Ophthalmology. 1984;91: 1253–1263.
Jabs DA, Mudun A, Dunn JP, et al. Episcleritis and scleritis: clinical features and treatment results. Ophthalmology. 2000;130:469–476.
Figure 3-2. A. Diffuse anterior scleritis is characterized by inflammation of the deep scleral and episcleral vessels, which have a dark red/violaceous hue. B. Diffuse anterior scleritis with inferotemporal thinning. Note the scleral injection and adjacent scleral thinning evidenced by the blue color of the underlying choroid.
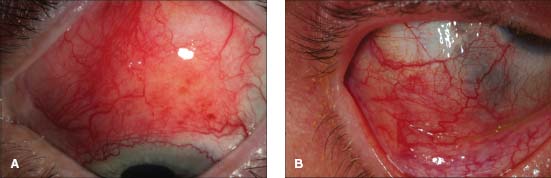
Figure 3-3. Nodular scleritis and peripheral ulcerative keratitis. The immobile nodule is surrounded by scleral injection. There is a focal, peripheral corneal opacity resulting from past episodes of peripheral ulcerative keratitis.
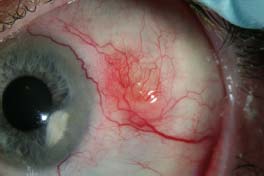
Figure 3-4. Necrotizing scleritis in a patient with Wegener’s granulomatosis with one suture remaining from a past scleral biopsy. Note the white, avascular patch of sclera with surrounding scleritis.
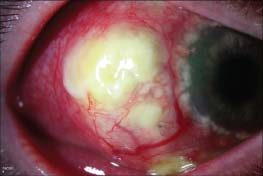
Figure 3-5. This elderly woman had poorly treated rheumatoid arthritis that caused painless scleral thinning through which the underlying blue choroid is visible (necrotizing scleritis without inflammation, or scleromalacia perforans). (Courtesy of Sunir J. Garg, MD.)
POSTERIOR SCLERITIS
It is an inflammatory disease of the sclera that begins posterior to the spiral of Tillaux/ora serrata and involves the posterior aspect of the eye.
Etiology and Epidemiology
 Posterior scleritis is more common in women. It has the lowest incidence of the various subtypes of scleritis; however, it is often underrecognized because of its many manifestations.
Posterior scleritis is more common in women. It has the lowest incidence of the various subtypes of scleritis; however, it is often underrecognized because of its many manifestations.
Symptoms
 Patients often present with an “achy, deep” pain, decreased vision, and redness.
Patients often present with an “achy, deep” pain, decreased vision, and redness.
Signs
 Unlike anterior scleritis, the vision is often affected.
Unlike anterior scleritis, the vision is often affected.
 Pain
Pain
 Elevated intraocular pressure, associated anterior scleritis, a swollen optic disc, choroidal folds, serous retinal detachment, and/or a subretinal mass or lesion (Fig. 3-6)
Elevated intraocular pressure, associated anterior scleritis, a swollen optic disc, choroidal folds, serous retinal detachment, and/or a subretinal mass or lesion (Fig. 3-6)
 T-sign on B-scan ultrasound (Fig. 3-7)
T-sign on B-scan ultrasound (Fig. 3-7)
Differential Diagnosis
 Choroidal tumors
Choroidal tumors
 Uveal effusion syndrome
Uveal effusion syndrome
 Rhegmatogenous retinal detachment
Rhegmatogenous retinal detachment
 Vogt-Koyanagi-Harada syndrome
Vogt-Koyanagi-Harada syndrome
 Central serous chorioretinopathy
Central serous chorioretinopathy
 Optic neuritis
Optic neuritis
 Masquerade syndromes (lymphoma, metastatic carcinoma, choroidal melanoma)
Masquerade syndromes (lymphoma, metastatic carcinoma, choroidal melanoma)
Diagnostic Evaluation
 Laboratory evaluation for an underlying systemic disease is warranted based on the results of a thorough history and review of systems.
Laboratory evaluation for an underlying systemic disease is warranted based on the results of a thorough history and review of systems.
 Rule out treatable infectious causes such as syphilis and tuberculosis (RPR, VDRL, FTA-Abs, PPD, Quantiferon)
Rule out treatable infectious causes such as syphilis and tuberculosis (RPR, VDRL, FTA-Abs, PPD, Quantiferon)
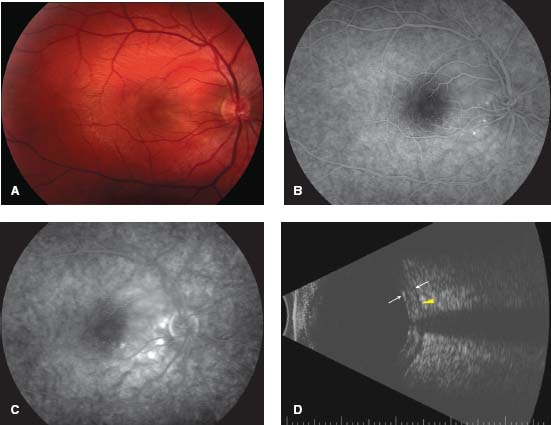
 B-scan: This is critical to establish the diagnosis. It demonstrates scleral wall thickness >2 mm in either a diffuse or nodular fashion. Classically, a T-sign that represents a sonographically empty space due to edema surrounding Tenon’s capsule and the optic nerve is seen.
B-scan: This is critical to establish the diagnosis. It demonstrates scleral wall thickness >2 mm in either a diffuse or nodular fashion. Classically, a T-sign that represents a sonographically empty space due to edema surrounding Tenon’s capsule and the optic nerve is seen.
 Fluorescein angiography can be used to rule out other causes such as Vogt-Koyanagi-Harada syndrome and sarcoidosis.
Fluorescein angiography can be used to rule out other causes such as Vogt-Koyanagi-Harada syndrome and sarcoidosis.
Treatment
 Most patients respond to oral NSAIDs; however, those with more severe chronic disease will require more aggressive systemic therapy with corticosteroids and/or immunosuppressive therapy.
Most patients respond to oral NSAIDs; however, those with more severe chronic disease will require more aggressive systemic therapy with corticosteroids and/or immunosuppressive therapy.
Prognosis
 The prognosis depends on the timeliness of treatment and severity of disease. Patients older than 50 years, patients with an associated systemic disease, and patients requiring more aggressive treatment have a greater risk of vision loss.
The prognosis depends on the timeliness of treatment and severity of disease. Patients older than 50 years, patients with an associated systemic disease, and patients requiring more aggressive treatment have a greater risk of vision loss.
REFERENCES
Benson WE. Posterior scleritis (review). Surv Ophthalmol. 1988;32(5):297–316.
Foster CS, Sainz de la Maza M. Clinical consideration of episcleritis and scleritis. In The Sclera. New York: Springer-Verlag; 1994:107.
Foster CS, Forstot SL, Wilson LA. Mortality rate in rheumatoid arthritis patients developing necrotizing scleritis or peripheral ulcerative keratitis: effects of immunosuppression. Ophthalmology. 1984;91:1253–1263.
Jabs DA, Mudun A, Dunn JP, et al. Episcleritis and scleritis: clinical features and treatment results. Ophthalmology. 2000;130:469–476.
McCluskey PJ, Watson PG, Lightman S, et al. Posterior scleritis: clinical features, systemic associations, and outcome in a large series of patients. Ophthalmology. 1999;106:2380–2386.
Figure 3-6. This patient has posterior scleritis. A. There are horizontal choroidal folds consistent with a mass (in this cases scleral inflammation) behind the globe. B. The fluorescein angiogram shows the choroidal striations. (Courtesy of William Benson, MD.)
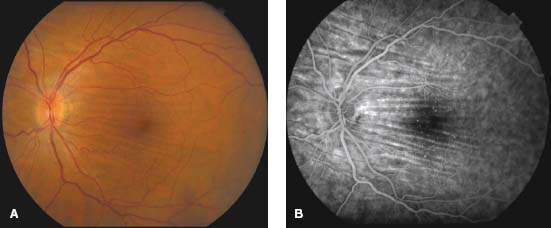
Figure 3-7. Posterior scleritis causes disruption of the normal choroidal circulation as well as restriction of scleral outflow through the vortex veins. This causes subsequent dysfunction of the retinal pigment epithelium. A. This can result in a serious retinal detachment. Fluorescein angiography can demonstrate pinpoint choroidal leakage (B) with pooling of dye in the late frames (C). D. B-scan ultrasonography demonstrates thickening of the sclera and choroid (arrows). Edema of Tenon’s capsule behind the globe, and along the optic nerve, creates the T-sign. (Courtesy of William Benson, MD, and Eliza Hoskins, MD.)
PHYLECTENULOSIS
S. R. Rathinam
Phlyctenular keratoconjunctivitis is a nonspecific, delayed hypersensitivity (type IV) reaction of the cornea and/or conjunctiva toward a variety of antigens.
Etiology and Epidemiology
 Phlyctenulosis is seen in the first two decades of life.
Phlyctenulosis is seen in the first two decades of life.
 It is more common in persons with poor personal hygiene and in those of lower socioeconomic status.
It is more common in persons with poor personal hygiene and in those of lower socioeconomic status.
 It is often associated with chronic meibomitis and chalazia.
It is often associated with chronic meibomitis and chalazia.
 Less commonly, it has been associated with pulmonary and extra pulmonary tuberculosis, staphylococcal infection, worm infestation, and focal sepsis.
Less commonly, it has been associated with pulmonary and extra pulmonary tuberculosis, staphylococcal infection, worm infestation, and focal sepsis.
Symptoms
 Lacrimation, photophobia, decreased vision, and blepharospasm
Lacrimation, photophobia, decreased vision, and blepharospasm
Signs
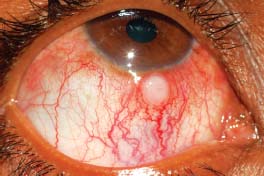
 Phlyctenulosis is a focal inflammatory disease characterized by an elevated, translucent nodule or vesicle with an ulcerated summit surrounded by a zone of hyperemia (Figs. 3-8 and 3-9).
Phlyctenulosis is a focal inflammatory disease characterized by an elevated, translucent nodule or vesicle with an ulcerated summit surrounded by a zone of hyperemia (Figs. 3-8 and 3-9).
 Often, conjunctival phlyctens are transient and asymptomatic.
Often, conjunctival phlyctens are transient and asymptomatic.
 However, larger phlyctens can result in frank pustular conjunctivitis.
However, larger phlyctens can result in frank pustular conjunctivitis.
 If the corneal phlycten migrates from its limbal origin to the central cornea, it is called fascicular keratitis, which causes severe vision loss.
If the corneal phlycten migrates from its limbal origin to the central cornea, it is called fascicular keratitis, which causes severe vision loss.
Differential Diagnosis
 Pingueculum
Pingueculum
 Episcleritis
Episcleritis
 Foreign-body granuloma
Foreign-body granuloma
 Scleral abscess
Scleral abscess
Diagnostic Evaluation
 Examination of the lid margin for blepharitis, meibomitis, and chalazia.
Examination of the lid margin for blepharitis, meibomitis, and chalazia.
 In countries such as India, consider:
In countries such as India, consider:
 Systemic workup for pulmonary/extrapulmonary tuberculosis
Systemic workup for pulmonary/extrapulmonary tuberculosis
 Screening for palpable lymph node to biopsy
Screening for palpable lymph node to biopsy
 Pulmonary radiological studies to rule out tuberculosis
Pulmonary radiological studies to rule out tuberculosis
 Stool examination for worm infestation
Stool examination for worm infestation
Treatment
 The primary treatment is good lid hygiene to eliminate staphylococcal lid infection; this includes warm moist compresses, lid scrubs with baby shampoo, and topical erythromycin eye ointment.
The primary treatment is good lid hygiene to eliminate staphylococcal lid infection; this includes warm moist compresses, lid scrubs with baby shampoo, and topical erythromycin eye ointment.
 If another systemic cause is found, treatment of the underlying disease is essential:
If another systemic cause is found, treatment of the underlying disease is essential:
 If due to a parasite, albendazole 400 mg/day can be used.
If due to a parasite, albendazole 400 mg/day can be used.
 In cases of tuberculosis, multidrug antitubercular treatment (rifampicin 10 mg/kg/day, isoniazid 5 mg/kg/day q.d. for 6 to 9 months, ethambutol 15 mg/kg/day, and pyrazinamide 25 to 30 mg/kg/day q.d. for first 2 months should be considered).
In cases of tuberculosis, multidrug antitubercular treatment (rifampicin 10 mg/kg/day, isoniazid 5 mg/kg/day q.d. for 6 to 9 months, ethambutol 15 mg/kg/day, and pyrazinamide 25 to 30 mg/kg/day q.d. for first 2 months should be considered).
 Prednisolone acetate 1% or dexamethasone 0.1% every two hours for 1 week followed by a slow taper hastens resolution.
Prednisolone acetate 1% or dexamethasone 0.1% every two hours for 1 week followed by a slow taper hastens resolution.
 In severe cases, or those that require longer-term steroid use, topical cyclosporine A 2% has been found to be effective.
In severe cases, or those that require longer-term steroid use, topical cyclosporine A 2% has been found to be effective.
Prognosis
 Conjunctival phlyctens heal without scarring and carry a good prognosis.
Conjunctival phlyctens heal without scarring and carry a good prognosis.
 Corneal phlyctens may cause stromal scarring, which can cause vision loss (Fig. 3-10).
Corneal phlyctens may cause stromal scarring, which can cause vision loss (Fig. 3-10).
 Recurrences are more common in patients with tuberculosis (Fig. 3-11).
Recurrences are more common in patients with tuberculosis (Fig. 3-11).
REFERENCE
Doan S, Gabison E, Gatinel D, et al. Topical cyclosporine A in severe steroid-dependent childhood phlyctenular keratoconjunctivitis. Am J Ophthalmol. 2006;141: 62–66.
Figure 3-8. This is a typical, moderate to severe phlyctenule with an elevated, translucent nodule with an ulcerated summit surrounded by a ring of hyperemia.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


