The Eye in Skeletal Disorders
Abraham Spierer
Tamara Wygnanski-Jaffe
Facial Deformity Syndromes
Mandibulofacial Dysostosis
Treacher Collins syndrome (TCS), mandibulofacial dysostosis or zygo auricular mandibular dysplasia is caused by malformations of structures formed from the first and second brachial arch, including the zygoma, ear, mandible, and eye.1 Clinical features characteristically include hypoplasia of the mandible, cleft palate, a malformed chin and zygoma, a wide range of ear deformities such as abnormalities of the pinnae, atresia of the external auditory canals, malformed ossicles, and middle ear and ocular abnormalities.2
TCS is an autosomal dominant disorder affecting 1/50,000 live births, with variable expressivity, ranging form perinatal death caused by airway problems to clinically undetectable cases. The responsible gene TCOF1 has been mapped to 5q32-33.27. The gene product is the protein, treacle. In most cases of TCS there is no family history.3
Over 50 disease causing mutations have been identified.4
Common ophthalmic findings include a defective lateral angle of the orbit, displacement of the superior orbit, causing the orbit to have an egg like appearance. Additional ocular findings include vision loss, amblyopia, refractive errors, strabismus, lid, and adnexal abnormalities.5 In addition, pseudocolobomas and colobomas of the lids, dermoids, and lipodermoids in variable locations are prevalent. Intraocular involvement is rare, but microphthalmos has been reported.6,7,8,9,10
Oculovertebral Dysplasia
Oculovertebral dysplasia (Weyers-Thier syndrome) is a unilateral variant of mandibulofacial dysostosis; it is rarer than the more typical forms of the Treacher Collins syndrome.11,12,13 It is characterized by three classes of deformities: those involving the face, those involving the spine and ribs, and those affecting the eye. The facial malformation results mostly from maxillary dysplasia. Thus, there is facial asymmetry, malformation of the superior alveoli, and dental malocclusion. On the affected side, other findings are a low-set and rudimentary external ear, an area of alopecia behind the affected ear, enlargement of the buccal commissure with unilateral macrostomia, and absence of the mandibular ramus. There may be faulty segmentation of the spine and rib anomalies. The hand on the involved side may be deformed.
Ocular findings are unilateral, and they include anophthalmos, cryptophthalmos, and microphthalmos. Weyers and Thier subdivided this syndrome into two types, that is, a typical unilateral mandibulofacial dysostosis and an oculoauricular form.
Pierre Robin Sequence
Pierre Rubin Sequence-Glossoptosis, micrognathia and cleft palate are usually accompanied by respiratory distress. The incidence of Pierre Rubin syndrome is 1:8,500 live births.14
Pierre Robin Sequence is associated with the deletion of 2q32.3-q33.2. The diagnostic triad consists of glossoptosis, micrognathia and cleft palate.
Mandibular growth occurs during the second month of pregnancy and coincides with closure of the palate. Because mandibular development in PRS is compromised, the tongue remains between the palatine process and the cleft of the palate persists.
Patients with PRS may have associated pediatric syndromes. PRS can be subdivided into syndromic and non syndromic forms, or PRS and PRS plus. In one third of the cases, PRS is part of a more complex syndrome, the most prevalent being Sticklers syndrome.15
The association between PRS and Stickler syndrome warrants an ophthalmic examination for every child with this syndrome. Associated ocular findings in Stickler syndrome include: high congenital myopia, retinal detachment, and an empty vitreous cavity. Congenital alacrima16 and microphthalmia have been reported as single cases.17 Nonsyndromic PRS was not found to recur in sibs.18
Newborns with PRS may experience airway problems requiring immediate attention.19 The micrognthia and glossoptosis result in abnormal in utero swallowing and consequently polyhydramnios. Prenatal diagnosis has been reported as early as 13 weeks of gestation.20
In mice, there is a relation between growth retardation of Meckel’s cartilage and malformation of the secondary palate as predicted by the RPS.21
Oculo auriculo-vertebral spectrum- Goldenhar’s Syndrome
Goldenhar’s syndrome is an etiologically heterogeneous disorder that expresses a continuum known as hemifacial microsomia. Gorlin concluded that significant overlap occurs between the clinical manifestations of hemifacial microsomia, Goldenhar syndrome and oculo auriculo vertebral spectrum and that no valid distinction between these three could be made.22 Consequently, Gorlin et al. adopted the term oculoauriculovertebral spectrum to describe this complex.23 The incidence of this syndrome was found by Gorlin to be 1/5600.23 Goldenhar syndrome causes first and second brachial arch anomalies. The first and second brachial arches contribute to craniofacial development, therefore interference with this procedure during 4 to 8 weeks of embryonic life results in maxillary, mandibular and ear abnormalities. The etiology is undetermined; however, there is an insult that affects the development in the fetus during the first trimester that results in mal development of the involved structures during embryogenesis.24
Although mostly sporadic, autosomal dominant and recessive modes of inheritance have been reported. Bilateral cases are mostly autosomal dominant. Discordant twins for Goldenhar’s syndrome have been reported; therefore, nongenetic factors play a role in the development of these features.25
Diagnosis of Goldenhar’s syndrome is based on abnormalities of the eye ear and vertebrae. Two systems need to be involved in order to classify a case as Goldenhar’s Syndrome. Eye abnormalities include lipomas, lipodermoids (Fig. 1), epibulbar dermoids, upper lid colobomas, and a case of Duane’s retraction syndrome have been reported with features of Goldenhar’s syndrome . The epibulbar dermoids originate in congenital changes of mesoderm and ectoderm and are usually located on the inferior temporal limbus of the cornea (Fig. 2). Additional reports found caruncle abnormalities26 motility disorders, and lacrimal drainage anomalies. Rare findings include: optic nerve hypoplasia, tortuous retinal vessels, and microphthalmia.
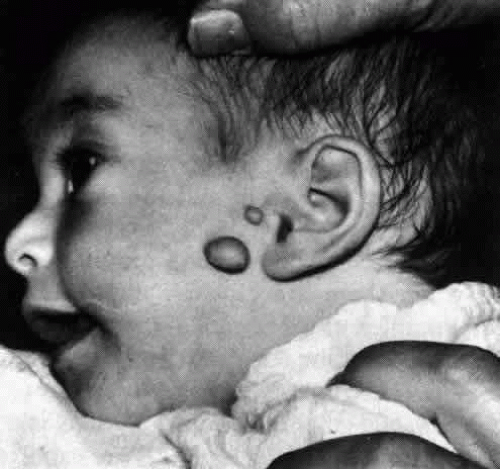 Figure 1. Lipodermoid presenting as subconjunctival mass adjacent to globe temporally and a dermoid tumor at the limbus. |
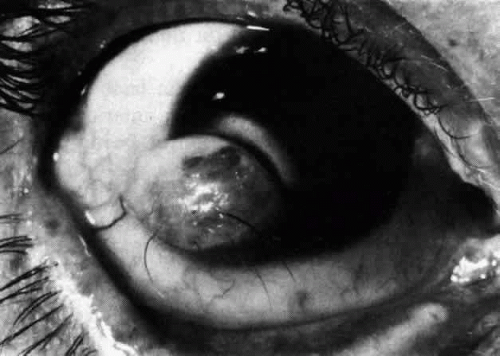 Figure 2. Epibulbar dermoid tumor at limbus, with prominent hairs protruding from it. |
Preauricular appendages are found in 70% of the patients (Fig. 3). Malformations of the outer ear or microtia are mandatory features for making the diagnosis.27 Changes in the vertebrae include butterfly vertebrae and hemivertebrae occurring at all levels of the spine. In addition to the facial structure cardiac, renal and skeletal anomalies may occur.28
 Figure 3. Preauricular appendages in a child with Goldenhar’s syndrome. |
Hallermann-Streiff (oculomandibulodyscephaly)
Hallermann-Streiff syndrome (HSS) is a syndrome of multiple congenital anomalies of the head and face, including dyscephaly, hypotrichosis, micrognathia and dental anomalies, nanism, cutaneous atrophy, hypotrichosis, and a beaked nose, which gives affected patients a birdlike appearance.29,30,31,32
The brow and parietal areas are prominent and the occiput is flat. The sutures and fontanelles remain open at least through infancy and frequently for many years. Size at birth is normal, although dwarfism develops during childhood. In the newborn, the tip of the nose is small and has a beaked appearance; older patients have a long thin nose and receding chin. There is frequently atrophy of the skin of the face. The hair is thin and sparse, with areas of baldness. The mouth and tongue are small, and the palate is high. Dental anomalies are seen in 50% to 80% of patients and include absence of teeth, natal teeth, enamel hypoplasia, supernumerary, and malformed teeth. Malocclusion is common. There may be anterior displacement of the temporomandibular joint. The ears are low set. The testicles are usually small and undescended. The joints are often hyperextensible, especially the knees. One in six patients shows mental retardation. Of the approximately 200 cases reported, familial occurrence has been infrequent. HSS is sporadic, but inheritance pattern is still questionable.32,33 No definite chromosomal abnormalities were identified.
Ocular findings include microphthalmos and microcornea, congenital cataracts, glaucoma, antimongoloid slant of the lids, blue scleras, pupillary membranes, nystagmus, strabismus, and retinal anomalies. Eyelashes and eyebrows often are sparse or absent. Orbits are small with reduced interorbital distance.
Progressive hemifacial atrophy: Parry-Romberg syndrome
This syndrome usually begins in the first or second decade of life in individuals that showed no morphological anomaly at birth. It was originally described by Parry (1825) and Henoch and Romberg (1846), and consists of slowly progressive atrophy of the skin, subcutaneous tissue, cartilage, and bone37 of essentially half the face, frequently accompanied by contralateral Jacksonian epilepsy, trigeminal neuralgia, and changes in the eyes and hair. Ophthalmic involvement is common with enophthalmos due to fat atrophy and subsequent changes in the palpebral fissures being the most frequently observed. Flattening of the maxilla, orbital floor defects, hypotony, pupillary and iris abnormalities, uveitis, scleral melting, and strabismus that may be restrictive or paralytic have also been reported.38
Pigmentary changes of the fundus have also been observed.39
The atrophy may begin with a scar of the forehead resembling a scar caused by a wound from a sword (en coup de sabre), which is caused by adherence of the skin to underlying tissues. Evidence of a Mendelian inheritance pattern is lacking.40
There is a reported case of a 16-year-old boy who developed facial changes at age 7 and had localized scleroderma on one leg and the trunk. The presence of antinuclear antibodies in his serum suggested that the Parry-Romberg syndrome may be a form of localized scleroderma. However, there is usually no sclerosis. A review of the literature does not support autosomal dominant inheritance. Nevertheless, there are theories that include a genetic disorder with low penetration, vascular diseases such as linear scleroderma, or a neurologic disorder. To date no effective therapy exists.
Orthodontic therapy can be undertaken to correct contraction of the jaw muscles. Enophthalmos may be alleviated by grafting of prosthesis in the orbit. Correction of the ocular alignment may be achieved by extraocular muscle surgery.41
Cranial and Craniofacial Deformity Syndromes
The Craniostenoses
The craniostenoses include a heterogeneous group of cranial anomalies characterized principally by the premature closure of one or more suture lines in the developing calvarium.42,43,44 The resulting defect is a function of45 which suture or sutures are closed,46 the time in development at which closure takes place, and47 the extent of closure. The clinical signs and symptoms produced by the craniostenoses depend on the degree of cerebral and foraminal compression that occurs as a result of the bony deformities.
The principal groupings include the following:
Oxycephaly (tower skull, turricephaly). This condition is due to premature closure of both the transverse and coronal sutures; the result is a high, wide head with a short anteroposterior diameter. Because the head is very high, the highest point being near the bregma, the name “tower skull” has been applied to this anomaly. It usually appears sporadically, although a familial incidence has been reported.
Scaphocephaly. This condition occurs secondary to premature closure of the sagittal suture, which results in a head with a very long anteroposterior diameter, a small transverse diameter, and a short height. Scaphocephaly may be inherited or sporadic, and when inherited it may be either autosomal dominant or recessive.
Plagiocephaly. This results from the unilateral closure of the coronal suture. The head appears asymmetrically enlarged on the side of the unfused suture.
Acrocephaly. The acrocephalic head is formed by premature closure of the anterior sagittal suture; this causes the posterior portion of the skull to bulge upward, leaving a sloping head with a very posterior summit.
Miscellaneous. Any cranial suture may close prematurely, resulting in the anomaly predicted by the anatomy of the skull.
The signs and symptoms produced by the craniostenoses are functions of the abnormal anatomy of the skull. Although prematurely closed sutures are closed at birth, their effect is not fully appreciated until the cranium starts to grow anomalously. The sutures are usually palpable and ridged. Diagnosis is readily apparent from the appearance of the patient, and radiologic studies are confirmatory. Occasionally, defects of the nasopharynx and palate are present concomitantly. Secondary cerebral atrophy and an increased incidence of retardation are probably results of cerebral compression. The symptoms of this group of disorders are mostly referable to the central nervous system. Increased intracranial pressure (occasionally intensified at puberty) may cause headaches, nausea, vomiting, and dizziness. Seizures are particularly common with oxycephaly. Other reported neurologic anomalies include: pyramidal tract dysfunction, anosmia, abducens palsy, trigeminal neuralgia, decreased hearing, and poor balance.
The eyes may be involved either as a direct consequence of the disease or by an associated anomaly. Optic atrophy may arise from any one of three causes. Primary optic atrophy may develop due to stretching of the optic nerves, which occurs when the brain is displaced by the cranial anomaly, or when the optic foramens are reduced in size by anomalous bone formation. Secondary optic atrophy may follow chronic papilledema. Exophthalmos may develop when the orbits are abnormally formed and shallow. Strabismus, especially exotropia, is common. Nystagmus, colobomas, and myelinated nerve fibers have also been reported.
The treatment of all these disorders is primarily surgical and is aimed at cosmetic repair of the cranial vault and decompression of the brain. Compression of the optic nerves may necessitate surgery in the area of the optic foramens.
Crouzon’s Syndrome
A rare deformity genetically related to Apert’s syndrome. The incidence of Crouzon’s syndrome is 1/25,000 live births. Crouzon’s and Apert’s syndrome are the most common causes of syndromic craniosynostosis. Crouzon’s syndrome is caused by a genetic aberration. It is usually sporadic and the risk for future children to have this syndrome is minimal. Nevertheless the risk of a Crouzon’s syndrome patient to pass the syndrome to their offspring is 50%-60% due to the dominant characteristics of this gene. Syndromic craniosynostosis has been found to be associated with mutations in the receptor gene for fibroblast growth factor (FGFR) Ig-like domain (FGFR2 effects periosteal fibroblasts bearing the FGFR2 receptor pro 253 Arg mutation). These mutations lead to premature ossification of the skull due to alternate signaling pathways.48,49,50,51
Children with Crouzon’s syndrome usually have normal intelligence although slightly limited intellectual capabilities have been reported.
The most prevalent characteristic of this syndrome is premature fusion of both coronal sutures resulting in a brachycephalic head. Additional features include fusion of the sagittal, as well as metopic and lamboid sutures. Due to protrusion of the forehead and eyebrows in addition to shallow orbits, exophthalmos usually characterizes these patients. Maxillary hypoplasia, hypertelorism, and a beaked nose are also typically found in this syndrome. Additional features include: short stature, spine abnormalities, a large protruding jaw, cleft palate, and misalignment of teeth (Fig. 4). Ophthalmic pathology is common. Visual impairment leading to acuity of 6/12 or less has been reported in up to 35% in one eye and 9% bilaterally. Visual impairment may be due to amblyopia, strabismus, optic atrophy, increased intracranial pressure causing chronic papilledema, refractive errors (both myopic and hypermetropic, anisometropia), ptosis, corneal exposure problems, and exophthalmos.52 Strabismus is many times the result of anomalous vertical horizontal and oblique muscle position within the orbit.
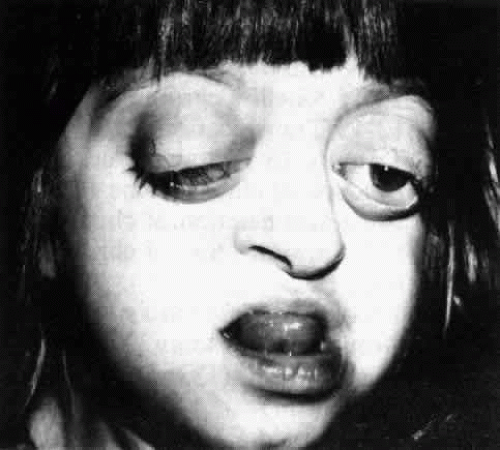 Figure 4. Typical facies in craniofacial dysostosis (Crouzon’s disease), with exophthalmos, hypertelorism, and exotropia. |
Treatment consists of multiple staged surgeries. The synostotic sutures are released to accommodate adequate cranial volume for growth of the brain. Repeated reshaping of the skull may be needed. Midfacial advancement and jaw surgery create adequate orbital volume and therefore reduce exophthalmos. Decompression around the optic foramen may be necessary in cases of chronic papilledema to relieve the pressure around the optic nerves, and special strabismus surgery may be needed due to malposition of the extra ocular muscles within the orbit.
Platybasia
Platybasia is abnormal flattening of the base of the skull occurring in several congenital disorders such as craniofacial anomalies, osteogenesis imperfecta, Arnold–Chiari malformation or in acquired diseases, such as Paget disease, osteomalacia, rickets, or trauma.53,54
When platybasia occurs with basilar invagination or upward migration of the cervical spine through the foramen magnum signs and symptoms of compression of the brainstem and upper cervical cord can result.57
Clinical manifestations of basilar invagination include symptoms of upper cervical cord compression and abnormal circulation of cerebrospinal fluid causing obstructive hydrocephalus.53,58
Isolated platybasia is of no clinical significance, and usually asymptomatic.
Basilar impression or invagination is the most prevalent malformation of the cranio-cervical junction, and causes cervical pain, cough, headache, lower cranial nerve palsies, corticospinal signs, hydrocephalus, cerebral dysfunction, and syringomyelia.59
Familial cases of platybasia have been reported.60
Nystagmus is the most characteristic feature and may be of any type. Especially localizing is the presence of downbeat nystagmus, which by itself frequently points to a lesion at the level of the foramen magnum. Absence of optokinetic nystagmus and asymmetric optokinetic nystagmus has also been noted, although explanation of the exact anatomic mechanism for this is lacking. Skew deviation may occur, probably as a result of distortion of the cerebellum and its connections, while a form of oculomotor dysmetria has also been described. Finally, chronic papilledema and secondary optic atrophy develop fairly frequently and may be present in either early, intermediate, or late stages of the disease.
The treatment of platybasia is surgical decompression of the posterior fossa.
Hypertelorism and Hypotelorism
Although hypertelorism was described in several textbooks dating back many centuries, it was Greig, in 1924, who proposed the current scientific explanation. This condition should be defined in rigid terms as a function of the bony structure of the skull and the angle formed by the medial walls of the two orbits (which are normally parallel).
Common facial features of the hypertelorism syndrome include: wide separation of the orbits; laterally displaced eyes with an exotropic strabismus; a broad, flat nasal bridge; and, frequently, a short upper lip. Intracranially, the sphenoid bone is often found to be deformed.61,62,63,64,65
Idiopathic hypertelorism as described by Greig probably is present in a small and unusual group of patients who have only the pure facial anomaly. This may be inherited as an autosomal dominant or recessive condition, has an unknown sex ratio, and is usually detected early in life.
Most cases of hypertelorism are associated with some generalized syndrome, which is presented under the following categories:
Chromosomal disorders
Craniofacial anomalies
Clefting syndromes
Systemic diseases affecting the face and head
Pseudohypertelorism may be due to anomalies of the canthi, nasal root, face, or orbits, which give the impression of increased separation of the orbits.
Hypotelorism is a rare facial anomaly usually occurring in conditions not compatible with survival.
The more common causes of these abnormalities are presented below.
Hypertelorism
Chromosomal disorders
Enlarged arms of the 4–5 chromosome
Short-arm deletion of chromosome 4
Short-arm deletion of chromosome 5 (cri-du-chat syndrome)
6–12 Translocation
6–12 Trisomy
Ring 13–15 (ring D)
Long-arm deletion of chromosome 13
13–15 Satellite chromosome
13–15 Translocation
Trisomy 13 (Patau’s syndrome)
Deletion of chromosome 16
Long-arm deletion of chromosome 18
Short-arm deletion of chromosome 18
Ring 18
Trisomy 18 (Edward’s syndrome)
Group G monosomy
Trisomy 21 (Down’s syndrome or mongolism)
The Schmid-Fraccaro syndrome
XO (Turner’s syndrome)
XXY (Klinefelter’s syndrome)
XXX (Superfemale)
Craniofacial anomalies
Acrocephalosyndactyly syndromes
Apert’s
Pfeiffer’s
Others
Cloverleaf skull (Kleeblattschädel syndrome)
Craniofacial dysostosis (Crouzon’s disease)
Craniometaphyseal dysplasia
Cranio-oculodental syndrome (Chotzen)
Craniostenoses
Oxycephaly (turricephaly, tower skull)
Acrocephaly
Scaphocephaly
Plagiocephaly
Others
Oculodentodigital dysplasia
The clefting syndromes
Cleft lip and palate, acrocephaly, radial aplasia, and absent digits
Cleft lip and palate, tetraphocomelia, and clitoral or penile enlargement syndrome (Appelt-Gerken-Lenz, Roberts syndrome)
Frontonasal dysplasia
Median cleft face syndrome
Systemic abnormalities
Aminopterin or amethopterin ingestion during pregnancy
Basal cell nevus syndrome
Cerebral gigantism
Cerebrohepatorenal syndrome (Bowen-Zellweger)
Chondrodystrophia calcificans congenita (Conradi’s syndrome)
Cretinism
de Lange syndrome
Dwarfing syndrome of Robinow-Silverman-Smith (fetal face syndrome)
Familial dysautonomia (Riley-Day syndrome)
Hallerman-Streiff syndrome
The happy puppet syndrome
Hypertelorism, acromegaloid features, and pectus carinatum
Infantile idiopathic hypercalcemia syndrome
Leprechaunism
Lissencephaly syndrome
Meckel’s syndrome (Gruber’s)
Multiple lentigines syndrome
Noonan’s syndrome (male Turner’s, Ullrich)
syndrome
Seckel-Virchow syndrome (bird-headed dwarfism)
Thymic agenesis (DiGeorge syndrome)
Apparent Hypertelorism
Divergence of the eye or eyes secondary to orbital disease
Polyostotic fibrous dysplasia of bone (Albright’s disease)
Thyroid ophthalmopathy
Any orbital tumor, pseudotumor
Any condition in which epicanthus or pseudoepican thus is present
Apparent orbital divergence due to flattened facies (syndrome of cleft palate, flattened facies, and multiple congenital dislocations)
Apparent hypertelorism due to a broad, flat nasal root
Chromosomal defects
Orofacial-digital syndrome
Otopalatodigital syndrome
Smith-Lemli-Opitz syndrome
Secondary to nondevelopmental nasal deformity
Congenital syphilis
Relapsing polychondritis
Trauma
Apparent separation of the orbits secondary to medial telecanthus
Acrocephalopolysyndactyly (Carpenter’s syndrome)
The hypertelorism-hypospadias syndrome (patients with this syndrome rarely have true hypertelorism)
Schwartz-Jampel syndrome (Aberfeld’s)Waardenburg syndrome
Hypotelorism
Cebocephalia
Craniotelencephalic dysplasia
Cyclopia
Ethmocephali
Holoprosencephaly (arrhinencephaly)
Holotelencephaly
Trisomy 13 (rare cases)
Weill-Marchesani syndrome
Generalized Skeletal Disorders
Apert’s Syndrome
First described by Apert in 1906, Apert’s syndrome (acrocephalosyndactyly) is a congenital anomaly affecting bones throughout the body. The main characteristics are premature fusion of the cranial sutures and syndactyly of the hands and feet (Fig. 5).42,66,67,68 Although most cases are sporadic, an autosomal dominant inheritance pattern has been demonstrated in some families. Birth prevalence of Apert syndrome was calculated to be 15.5/1,000,000 births. Males and females are affected equally. Some genetic studies have implicated increasing parental (especially paternal) age as a related factor. The condition has been reported in nearly all races, and the intelligence of the affected individuals is usually normal.
 Figure 5. Syndactyly of the hand in Apert syndrome. (Courtesy Moshe Frydman MD, Sheba Medical Center). |
Apert’s syndrome results from mutations of the fibroblast growth-factor receptor 2 gene (FGFR2) located at chromosome 10.70
The cranial malformation is principally a premature synostosis of the coronal suture and the hypoplasia of the mid base of the skull and the central face. The bones involved in the latter comprise the sphenoethmo-maxillary complex. The sutural synostosis leads to a characteristic oxycephalic skull deformity in which there is an inconspicuous occiput and a high point anterior to the bregma. The forehead is vertically oriented with no slope, and the anteroposterior diameter of the skull is greatly shortened. Neurologic disturbance is a rare consequence of the cranial malformation. These patients are of normal intelligence unless there is neurologic insult. A broad, depressed nasal bridge and relative prognathism are intrinsic parts of the facial anomaly; posterior cleft palate is occasionally reported.
Other skeletal anomalies most frequently involve the vertebral bodies, the digits, and the limb girdles. A fusion of the fingers is an integral part of the syndrome and characteristically involves the second through fourth digits. The presence of a single fused fingernail is especially characteristic, while involvement of the first and fifth digits is seen occasionally. The syndactyly involving the lower extremities is more likely to include a soft-tissue union of the affected members than that of the upper extremities. The vertebral bodies exhibit varying degrees of fusion, which tends to increase with advancing age. Finally, varying bony abnormalities of the shoulder and pelvic girdles may occur, which result in decreased range of motion along with pain and difficulty in active and passive movements.
Visceral anomalies have been described in this syndrome. In one series,71 cardiovascular anomalies were found in 10% of the patients and genitourinary anomalies occurred in 9.6%. Less frequent anomalies were found in the respiratory system (1.5%) and gastrointestinal system (1.5%).
The ocular findings are those secondary to the frequently present cranial anomalies and those which occur in relation to, but not as a direct consequence of, the bony malformations. An antimongoloid palpebral slant with the outer canthi measurably lower than the inner canthi is common. Strabismus is found and, rarely, congenital absence of the superior and inferior rectus muscles as well as the inferior and superior oblique muscles. Proptosis secondary to an anatomically shallow orbit may lead to severe exposure keratitis, corneal ulcers, and scarring.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


