Structure and Function of the Retinal Pigment Epithelium
Gerald B. Grunwald
The retinal pigment epithelium (RPE) occupies a functionally critical location in the human eye, sandwiched between the neural retina (NR) and the choroid. At first glance, the RPE appears strikingly simple and homogeneous in histological organization, presenting as a simple epithelial monolayer of pigmented, hexagonally packed cuboidal cells. However, this apparent simplicity is deceptive, because the RPE actually performs a wide variety of functions that are critical during the embryonic development of the retina as well as throughout adult life to maintain normal visual function (see Table 1). These functions include absorption of stray light to enhance visual acuity, protection against toxic and oxidative damage, formation of the blood–retinal barrier, selective transport of substances to and from the neural retina, phagocytosis of shed photoreceptor outer segments, elimination of waste products, and processing of vitamin A metabolites in the visual cycle. The purpose of this review is to provide an overview of the structure and function of the RPE, with an emphasis on its cellular, molecular, and developmental biology. Given the critical role of the RPE in retinal function, and the growing recognition of RPE dysfunction as a cause of ocular disease, a greater understanding of the fundamental cellular and molecular biology of RPE will be important to take full advantage of the emerging prospects for treatment of these diseases through physi-ologic and genetic modulation of RPE function and through RPE cell transplantation. The existing literature on the biology of the RPE cell is vast, and this chapter summarizes fundamental aspects as well as current advances in our understanding of the RPE in the decade since the last version of this chapter was written. This previous chapter may be referred to for additional background and its excellent and detailed review of RPE cytology.1 In addition, the reader is directed to additional relevant current chapters of this compendium.
Historically, by the mid-19th century, the RPE was recognized as a distinct ocular tissue within the eye as a result of advances in histology and microscopy, as well as an appreciation of its embryonic origin.2 Notable for its melanin content and resulting deep pigmentation, this property, coupled with the location of the RPE just posterior to the retina, at first suggested a primary function in absorption of stray light that had not been absorbed by the retina, which would otherwise result in degradation of the visual image caused by reflection and scattering within the eye. However, even 19th century clinical observations had already linked detachment of the neural retina from the RPE to loss of visual function, and suggested that the RPE possessed a more active and vital role in visual physiology. Subsequent clinical findings, combined with extensive in vitro cellular and molecular analyses, and in vivo animal model studies, have clearly identified the RPE as a physiologically complex tissue with a variety of functions that support the visual process. Enhanced understanding of the cellular and molecular basis of retinal disorders has identified RPE dysfunction as playing either a direct primary role, or an indirect secondary role through interaction with the NR, in ocular diseases such as age-related macular degeneration (ARMD), proliferative vitreoretinopathy (PVR), retinitis pigmentosa (RP), Stargardt disease, Leber’s congenital amaurosis, and congenital hyperplasia of the RPE (CHRPE), which are discussed further.
TABLE 1. Summary of Structural and Functional Specializations of the RPE | |
|---|---|
|
EMBRYONIC DEVELOPMENT AND CONGENITAL DISORDERS OF THE RPE
The embryonic origin of the RPE, along with the neural retina, can be traced back to formation of the neural tube during neurulation, with subsequent formation of the optic vesicles as outpouchings of the diencephalic region of the primitive brain. Invagination of the optic vesicles results in formation of a two-layered optic cup, with the RPE derived from the outer layer and the neural retina derived from the inner layer (Fig. 1). Thus, the RPE is a neuroepithelial derivative that becomes highly specialized in structure and function as an epithelial monolayer with unique properties.3 Because of the topological relationship of the nascent RPE and NR tissues during formation of the optic cup and optic vesicle, the RPE comes to lie adjacent and closely apposed to the NR, but with these two layers facing each other in an apical-to-apical fashion with respect to cellular polarity. This topographic relationship thus permits the intimate interaction of the apical microvilli of RPE cells with the apical outer segments of retinal rod and cone photoreceptors found in the mature retina (Figs. 2 and 3). On a gross level, the orientation of the RPE is such that its basal epithelial surface is oriented toward the outside of the eye, whereas the apical surface faces inward toward the vitreous chamber. Although intimately associated, the RPE and NR do not truly fuse to form a single coherent tissue, and although the original lumen of the optic vesicle is greatly reduced, this potential space between their apical surfaces persists as the intraretinal space (or subretinal space if expanded experimentally, surgically, or pathologically). As a consequence of the persistence of this potential space, retinal detachment may result from traumatic injury, with subsequent loss of visual function unless surgical re-attachment is made. However, the RPE and NR nevertheless normally remain intimately associated through RPE microvilli-NR-outer segment interdigitation, as well as by the presence of a specialized extracellular matrix known as the interphotoreceptor matrix.4
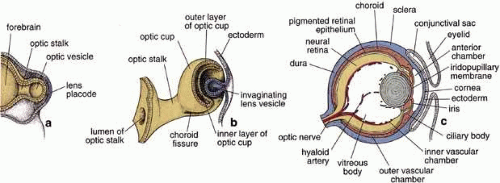 Fig. 1 Schematic drawing illustrating the development of the eye. Forebrain and developing optic vesicles as seen in a 4-mm embryo (a). Bi-layered optic cup and invaginating lens vesicle as seen in a 7.5-mm embryo (b). The optic stalk connects the developing eye to the brain. The eye as seen in a 15-week fetus (c). All the layers of the eye are established, and the hyaloid artery traverses the vitreous body from the optic disc to the posterior surface of the lens. (Modified from Mann IC, Th Development of the Human Eye. New York, Grune and Stratton, 1974, and reproduced from Ross et al. 2003) |
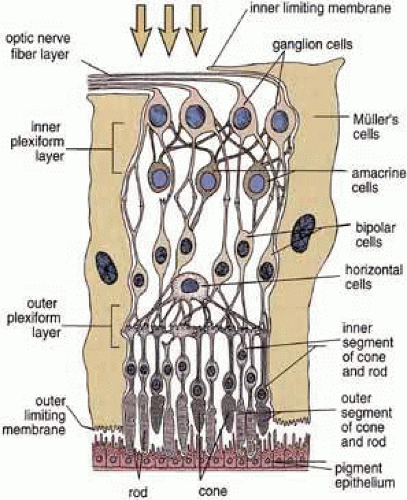 Fig. 2 Schematic drawing of the layers of the retina. The interrelationship of the neurons is indicated. Light enters the retina and passes through the inner layers before reaching the photoreceptors of the rods and cones that are closely associated with the retinal pigment epithelium. Reproduced from Ross et al. (2003). |
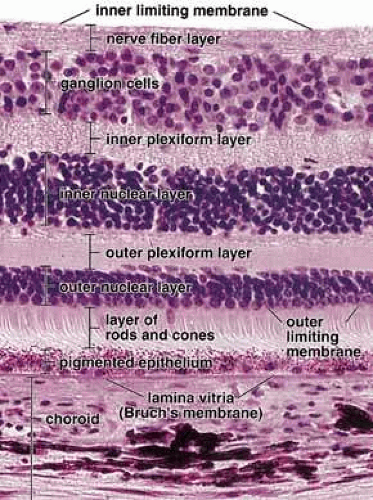 Fig. 3 Photomicrograph of a human retina. On the basis of histologic features, the retina can be divided into 10 layers as indicated on this photomicrograph. Note that Bruch’s membrane (lamina vitrea) separates the inner layer of the vascular coat (choroid) from the pigment epithelium, ×440. Reproduced from Ross et al. (2003). |
The focus of this review is the RPE proper, i.e., that portion of the RPE in association with the NR. It should be noted that the optic cup is double-layered throughout its extent to its most anterior margin, and that specializa- tions of this anterior margin give rise, through interaction with surrounding mesenchymal tissues, to the ciliary body and iris. Thus, the RPE also makes a contribution to these structures as the RPE of the ciliary body and its processes, as well as the RPE of the iris, with their own unique secretory and contractile properties.5
Proper differentiation of the RPE from the outer layer of the optic cup is dependent on signals received from surrounding tissues, including both the embryonic surface ectoderm as well as the loose extraocular mesenchymal tissue that fills the space between the presumptive RPE and the ectoderm. One of the specific signals that triggers this differentiation is the growth factor activin, a member of the TGFβ family.6 Such signals activate specific changes in gene expression, and advances in our understanding of the genetics of ocular development and disease have led to the identification of several genes that are required for normal development of the RPE as well as a number of human mutations that are now known to be associated with congenital anomalies of the RPE. For example, mutations of the gene encoding microphthalmia-associated transcription factor (MITF) result in forms of Waardenburg syndrome and Tietz syndrome, characterized by hypopigmentation and deafness. Although a variety of isoforms of MITF occur in a number of tissue types, MITF-A is particularly associated with the RPE.7 Another transcription factor controlling RPE differentiation is Otx2.8 MITF and Otx2 both appear to be required for maintenance of several functions of RPE cells, including proliferation, differentiation, and survival. Recent research has also identified several potential regulatory and patterning genes that establish the RPE phenotype, although these also affect additional aspects of ocular development including the NR, lens, and cornea. These include Pax2 and Pax6, in whose absence the external layer of the optic cup tissues develop into neural retina rather than RPE, or when overexpressed can induce neighboring tissues to develop RPE-like properties.9 Thus, differentiation of the RPE, as with other specific tissues, results from a complex interplay of genetic regulation and signaling pathways whose correct balance is required for proper embryonic development.
Some abnormalities of the RPE, while themselves sometimes relatively benign and not necessarily presenting an immediate threat to visual function, may nevertheless reflect other systemic problems. For example, congenital hyperplasia of the RPE (CHRPE) is recognized on fundus examination as a flat hyperpigmented region of the retina. Although isolated nonmalignant hamartomas of the RPE may occur, with little apparent effect on vision,10 it is important to distinguish such a finding from a choroidal melanoma. Furthermore, CHRPE has been associated as part of a syndrome including familial adenomatous polyposis (FAP), resulting from a mutation in the adenomatous polyposis coli (APC) gene. Although generally thought to represent a benign RPE hyperplasia with little effect on the adjacent retina, in this case CHRPE is associated with FAP and the extensive formation of colon polyps usually progressing to colon cancer.11 Furthermore, recent studies have indicated that CHRPE itself may progress to adenocarcinoma.12,13 Given this and the further association of altered RPE pigmentation with a variety of ocular diseases as discussed, the RPE can serve as an accessible sentinel for detection of ocular and systemic diseases, which may be further distinguished after ophthalmoscopic examination with fluorescein angiography and other diagnostic tests.14
CELLULAR ORGANIZATION OF THE RPE
The histological appearance of the mature RPE proper (that associated with the NR) is of a simple cuboidal polarized epithelium.15 When viewed en face, as seen in an explanted intact sheet of RPE, the cells generally appear as tightly adherent cells with hexagonal packing. RPE cells possess a characteristic cytological appearance and organelle distribution (Fig. 4). In RPE cells, mitochondria are located basally, beneath the nucleus, and close to the basal infoldings of the plasma membrane. The cells contain numerous catalase-containing microperoxisomes that function in the conversion of hydrogen peroxide to water. The RPE cell cytoplasm contains mainly smooth, and relatively little rough, endoplasmic reticulum, a characteristic of cells actively involved in lipid metabolism.15 RPE cells possess a Golgi complex, an organelle in which newly synthesized molecules are sorted, modified, and targeted to appropriate sites in the cell, a function critical for maintenance of RPE cell polarity. The Golgi complex of RPE cells is small and often scarcely distinguishable from the other tubules and vesicles of the endoplasmic reticulum. Lipid droplets (homogeneous-appearing spheres 0.5 to 1 μm in diameter with no limiting membrane) are seen rarely in primate pigment epithelial cells but are common in amphibian and rat retinas, where they have been shown to be a normal site of vitamin A storage. There are approximately 5 million RPE cells in the human eye, and during development the density of pigment epithelial cells increases steadily in the macular area, gradually reaching a stable level 6 months after birth. In contrast, near the ora serrata, cell density starts at high levels and decreases rapidly through the first postnatal year and more gradually thereafter. Furthermore, there is some concomitant variation in the dimensions of RPE cells depending on the location in the eye, and further variation may occur with aging.16,17 In the macular region of the adult eye the cells are tall (14–16 μm) and narrow (10–14 μm), whereas toward the periphery they become significantly flatter and wider, such that at the ora serrata RPE cells may be 60-μm-wide (Fig. 5). After age 60, RPE cells throughout most of the retina become shorter, broader, and generally demonstrate a more variable morphology, with macular RPE cells increasing in height with advancing age. However, after age 90, when there has been cell loss, even macular RPE cells become wider and flatter. While these events represent generally slow responses to age-related intrinsic and extrinsic changes, RPE cells both in vitro and in vivo can exhibit rapid and wide-ranging phenotypic variation including epithelial-mesenchymal transformation and transdifferentiation.18,19 Such a capacity for plasticity may represent a necessary and beneficial ability to respond to disease and injury, and indeed differentiated mammalian RPE cells remain capable of cell division and wound healing.20 However, there is limited capacity for extensive repair, such as after damage of the deeper layers of Bruch’s membrane, which often leads to scarring and lack of normal re-pigmentation.21 Furthermore, the normal program of RPE wound healing may be subverted by events such as exposure to vitreous and serum after rhegmatogenous retinal detachment, possibly resulting in the aberrant wound healing response and subsequent scarring seen in PVR.22,23 Thus, further elucidation of the molecular mechanisms underlying the phenotypic variability of RPE cells may provide important insights leading to therapeutic interventions in such circumstances.
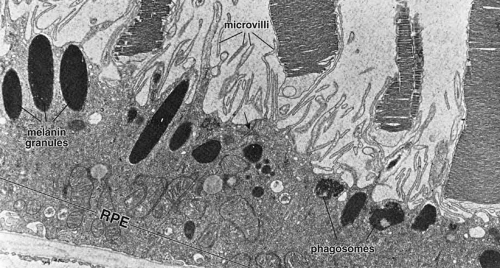 Fig. 4 Electron micrograph of the retinal pigment epithelium in association with the outer segments of rods and cones. Retinal pigment epithelium (RPE) contains numerous elongated melanin granules that are aggregated in the apical portion of the cell, where the microvilli extend from the surface toward the outer segments of the rod and cone cells. The retina pigment epithelial cells contain numerous mitochondria and phagosomes. The arrow indicates the location of the junction complex between two adjacent cells, ×20,000. (Courtesy of Dr. Toichiro Kuwabara and reproduced from Ross et al. 2003) |
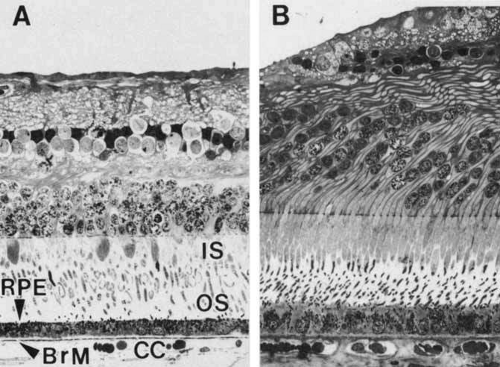 Fig. 5 Light micrographs of two areas of the same human retina sectioned in a transverse plane and aligned at Bruch’s membrane (BrM), demonstrating topographic variation. A. Equatorial retina. Cuboidal retinal pigment epithelial (RPE) cells, with basally located nuclei, form a continuous monolayer that marks the outermost limit of the retina. Each cell extends apical processes into the interphotoreceptor space, which interdigitate with the apical processes (the inner and outer segments [IS, OS]) of the photoreceptor cells of the inverted neurosensory retina. The basal surface of the RPE rests on Bruch’s membrane. Capillaries of the choroid (choriocapillaris, [CC]) abut the outer side of Bruch’s membrane. B. Fovea. Retinal pigment epithelial cells are taller and narrower. Bruch’s membrane is thicker (×320). |
ESTABLISHMENT OF RPE CELL POLARITY
Knowledge of the important functional role of the RPE cell membrane, and its molecular composition, have increased enormously with progress in the field of molecular cell biology, with substantial progress in our understanding of how the RPE regulates cell–cell and cell–matrix adhesion to maintain normal tissue integrity and polarity, and the associated barrier, transport, and secretory functions. Vectorial transport, a principal function of epithelia, depends on the polar distribution of plasma membrane constituents, and as in other polarized epithelial cells, the RPE surface is divided into apical and basolateral domains, each of which is discussed in detail. Both intrinsic and extrinsic signals contribute to the established polarity of cells, including RPE.24 Principle among these are the endogenous sorting mechanisms encoded into the polypeptide sequences of specific proteins and recognized by the Golgi apparatus, endoplasmic reticulum, and associated intracellular transport machinery, in concert with the morphogenetic signaling potential of cell–cell and cell–matrix interactions that guide a cell into appropriate relationships with their neighbors. For RPE cells, such guidance mechanisms are required for the polarized expression of ion pumps such as the Na+-K+ ATPase, which in RPE, as opposed to most epithelial cells, is localized apically, and carrier proteins such as the monocarboxylate transporters.3,25,26,27 Without such domain-specific localization patterns, the vectorial metabolic pumping and transport functions of the RPE for ions, water, visual cycle intermediates, nutrients, waste products, and macromolecular components of the IPM and Bruch’s membrane would not function properly. Experimental evidence suggests that the mechanisms that result in the proper placement and/or retention of RPE membrane proteins may include age-dependent changes during development, inductive interactions from neighboring tissues such as the NR, cell-junction-complex-mediated signaling by cadherin cell adhesion molecules, quantitative differences in membrane surface area of different subcellular compartments, random delivery followed by selective retention, and guidance by chaperone proteins.24,25,26,27,28,29 Disturbance in these mechanisms may result in the aberrant transport or accumulation of materials that characterize certain RPE-related retinal dystrophies, as discussed further.
APICAL MEMBRANE SPECIALIZATIONS OF THE RPE
As already discussed, the topography of retinal development results in the intimate association of RPE apical microvilli with rod and cone photoreceptor outer segments across the intraretinal space (Figs. 4 and 6). These are bound together by the interphotoreceptor matrix (IPM), a network of proteins and proteoglycans containing a variety of enzymes, growth factors, carrier proteins, and other constituents.4 A number of constituents of this matrix have been localized in three distinct patterns, such as those that demonstrate rod- and cone-specific localization, those with an apical-to-basal heterogeneity, and others with a more homogeneous distribution. When there is a neurosensory retinal detachment, the potential interphotoreceptor space expands as fluid accumulates to form what is clinically referred to as the subretinal space, and the photoreceptors, now deprived of their supportive RPE functions, will degenerate if re-attachment is not effective. To maintain normal retinal attachment, RPE cells develop long slender apical microvilli of 5 to 7μm in length, forming sheaths that appear to participate in phagocytosis of outer segments. Villous processes surrounding rods contain smooth endoplasmic reticulum, ribosomes, melanin granules, and actin filaments. Villous processes that surround extrafoveal cones are usually devoid of intracellular organelles except for pigment granules. Despite their intimate relationship, no junctional attachments have been found between the RPE apical processes and the photoreceptor outer segments, although several molecular mechanisms forming the basis of this recognition and adhesion have been proposed, as discussed later.
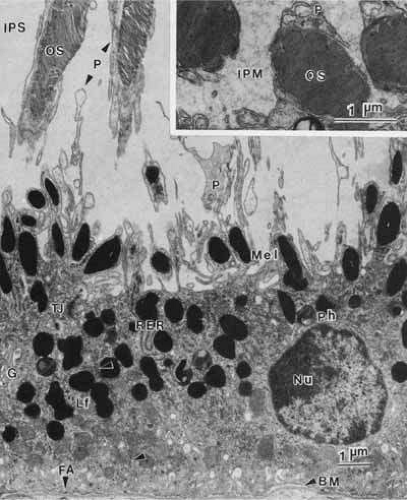 Fig. 6 Transmission electron micrograph of retinal pigment epithelium in a transverse plane. The basally located nucleus contains a nucleolus (Nu), loose euchromatin and marginated dense heterochromatin. The basal surface of the cell, marked by many infoldings of the plasma membrane, rests on a basement membrane (BM). The apical surface has long processes (P) extending into the interphotoreceptor space (IPS). These processes envelope the distal part of photoreceptor outer segments (OS). The junctional complex (TJ) is the site of attachment between adjacent epithelial cells (arrows). The retinal pigment epithelial cell contains two classes of pigments: elliptical melanin granules (Mel), located apically, and round or figure-eight lipofuscin granules (Lf), located more basally. Phagolysosomes (Ph) and other secondary lysosomes are seen. The cytoplasm contains principally smooth endoplasmic reticulum and a few lamellar arrays of rough endoplasmic reticulum (RER), which is usually located in the apical region. The paranuclear Golgi apparatus (G) is generally inconspicuous. Numerous mitochondria, free ribosomes, and microperoxisomes lie near the infoldings of the basal plasma membrane. Focal adhesions (FA) are special sites connecting the cell to its basement membrane. This specimen is from a 31-year-old woman (×10,500). Inset. Cross-section of outer segments (OS) near the apex of a retinal pigment epithelial cell showing the encircling apical process (P). Note the presence of interphotoreceptor matrix (IPM) in this specimen from a 26-year-old man (×23,000). |
As previously mentioned, the tight junctions of the RPE contribute to formation of the blood–retinal barrier and also help to establish the compartmentalization required to maintain the unique microenvironment of the IPM. In part this involves control of the ionic milieu required for phototransduction and its component dark current in photoreceptors. The apical membrane contains ion channels and transport molecules involved in fluid movement from the retina to the choroid.30 As already alluded to, unlike most transporting epithelia, which have Na+-K+ ATPase located in the basolateral plasma membrane near the energy-producing mitochondria, the RPE has this enzyme in the apical membrane. Na+-K+ ATPase helps regulate extracellular potassium levels and fluid fluxes that contribute to the adhesion of the neurosensory retina. RPE cells make an important contribution to this via regulation of K+ transport mediated by the KIR family of inwardly-rectifying K+ channel proteins, specific isoforms of which are expressed in the RPE and are localized along the cell surface membranes of the apical microvilli.31 Another important apical specialization of RPE cells is the localization of Na+K+/Cl– cotransport proteins that, in concert with additional basal Cl– channels (see later), regulate the chloride flux that appears to be the major determinant of net fluid transport across the RPE.30 This net vectorial transport of fluid from retina to choroid helps to maintain RPE/NR adhesion, and water-conducting aquaphorin membrane channels have been identified on the apical surface of RPE cells that facilitate this process.32 Additionally, apical localization of the monocarboxylate transporter MCT1 isoform may help regulate pH and osmolarity in the intraretinal space (again, see later for a basal membrane counterpart).33 Finally, another important specialization of the apical membranes of RPE cells is the presence of receptors that mediate binding and phagocytosis of shed photoreceptor outer segment membranes, and transport of vitamin A metabolites in the visual cycle, which is described further.
LATERAL SURFACE CELL-ADHESIVE INTERACTIONS OF THE RPE
The epithelial integrity of the RPE, as with all epithelial tissues, is critically dependent on lateral cell-cell interactions mediated by a variety of specialized intercellular junctions. The lateral cell membranes of the RPE have relatively flat surfaces, in contrast to the highly convoluted apical and basal surfaces (Figs. 7 and 8). As in typical epithelial cells, the cell-cell junctions of RPE cells form a classical “junctional complex” along the lateral cell membranes, and are arranged in the apical to basal direction as: (1) tight junctions or zonula occludens; (2) adherens junctions or zonula adherens; and (3) desmosomes or macula adherens34,35 (Figs. 4 and 9). However, the latter are not observed as prominently as in many other epithelia, and although desmosomes are observed in human RPE, there is some species-specificity of appearance.36 In addition, a fourth type of cell–cell junction, the gap junction or communicating junction, is also found in RPE cells. While originally classified on the basis of their morphologically distinct electron microscopic appearance, the molecular composition of these cell junctions has now been well characterized. Furthermore, these junctions are recognized to function as more than passive intercellular glues that bind RPE cells together, but in addition they function as loci of bi-directional signaling, integrating the cytoskeleton, intracellular metabolism and gene expression with the extracellular milieu (Figs. 9 and 10).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


