Chapter 14 Structure and Function of Rod and Cone Photoreceptors
Introduction
Our visual experience is initiated by rod and cone photoreceptors in the retina. The human eye contains over 100 million rods and about 6 million cones, which are located within the outer nuclear layer of the retina and allow our visual experience to extend over 12 orders of magnitude in light intensity by splitting this range. Rod photoreceptors mediate vision under conditions of dim illumination, and allow our visual system to reach the limit imposed by the absorption of single photons.1,2 Cones are less sensitive by ~100-fold, but their tremendous capacity for adaptation allows them to encode light intensities in the brightest of days.3,4 Over the last decade, our understanding of structure and function of these cells has increased dramatically. Over 150 genes have been cloned or linked to retinal diseases, and, surprisingly, as many as half of these genes are specifically expressed or highly enriched in the photoreceptor cells. Discovery of the molecular constituents of the rods and cones is progressing at an increasing rate, particularly enhanced by the availability of “complete” genomic sequences for both human and mouse. While photoreceptor genes can be identified through association with a retinal disease (linkage), information about their function does not accompany their identification.5 Basic science research must then be undertaken to explain their role in both the normal, healthy photoreceptor as well as in photoreceptor diseases.
Though much of what we know about the structure and function of photoreceptor cells has come from studying animal models of inherited blindness,6,7 more recently, transgenic and knockout animal technologies have established themselves as powerful tools for understanding function and studying disease. After new photoreceptor genes are identified from patients, mutant animals can be engineered to emulate human photoreceptor pathologies. Before these molecular technologies were available, most data about retinal disease were gleaned from the rare “informative” patient or donor retina that had surviving photoreceptors to examine.8 Other major sources of information were studies of animal models that occurred through inbreeding or random inheritance of mutations in photoreceptor genes. Examples of these are the Irish Setter dog,9 the Briard dog,10 the Abyssinian cat,11 the Royal College of Surgeons (RCS) rat,12 and the rd13,14 and rds15,16 mouse models. Now, the ability to engineer transgenic animals has made the search for “informative” patients and naturally occurring animal models less acute, since any single gene of interest can be introduced17–19 or removed20–24 from the photoreceptor. However, the most instructive examples of structure–function relationships occur when there are patients and animal models with analogous mutation(s) and/or disease phenotype.
Photoreceptor fundamentals
The rod and cone photoreceptors are specialized sensory neurons that contain the protein machinery necessary to convert incident light into a signal that can be interpreted by the nervous system. Rod photoreceptors are more numerous than cones in most mammalian retina, and are highly sensitive. In the fully dark-adapted state, rods can reliably report the absorption of single photons to the retinal output, and they permit our scotopic, or night, vision. Cone photoreceptors are morphologically and functionally distinct from rods and express several types of visual pigments, or opsins, whose spectral sensitivity varies based on the cone’s subtype. In humans, three classes of cones confer robust color vision: S, M, and L cones (see Chapter 10, Color vision and night vision). Cones are also less sensitive than rods and generate light responses that are temporally briefer. This allows cones to mediate our photopic, or day, vision with improved temporal resolution. The concerted action of these two types of photoreceptors, and the retinal circuitry that carries their signals to the retinal output, ultimately underlie our rich visual experience.
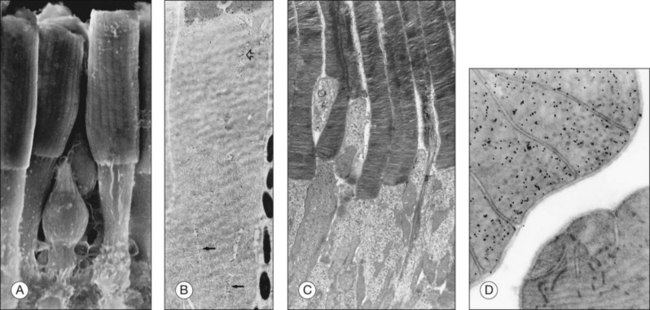
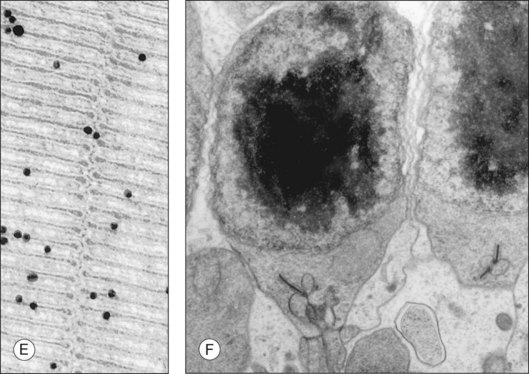
Both rod and cone photoreceptors are highly polarized elongated cells that can be described as having four subcellular compartments: the outer segment (OS), the inner segment (IS), the nucleus, and the synaptic terminal (Figs 14.1 and 14.2). The OS is where photons are captured and activation of the phototransduction cascade begins. The IS lies immediately proximal to the OS, and contains the cell’s protein synthesis (Golgi apparatus and endoplasmic reticulum) and metabolic (mitochondria) machinery. Light-evoked signals are relayed passively down the photoreceptor axon (up to 75 µm long) to the synaptic terminals in the outer plexiform layer. The structure of photoreceptor terminals is unique in the nervous system, as they contain a specialized structure called a ribbon that facilitates the release of the excitatory neurotransmitter glutamate on to second-order retinal neurons (bipolar and horizontal cells). Thus, the photoreceptor cells transduce the sensory stimulus, light, and pass on a signal to retinal circuits that carry this information to higher visual centers.
Photoreceptor outer-segment structure
The OS compartment contains all components necessary for phototransduction, which is a set of biochemical reactions that convert photon capture to a change in a cationic current at the plasma membrane. Rods have cylindrical OS about 1.3 µm in diameter and length that ranges from 25 to 45 µm,25–28 that depends on numerous factors, including the time of day, the light intensity, their location in the retina, and the animal species. The cone OS is shorter, typically half the length of rods, with a larger diameter at the base that gradually tapers towards the tip (Fig. 14.2). Mouse rod OS contains an average of 810 membranous discs, which occupy two-thirds of the volume of the OS.29 The majority of the protein on these discs is rod opsin (or rhodopsin when the opsin is bound to its chromophore, 11-cis retinal). The density of rhodopsin on the disc membrane has been measured to be about 24 000 molecules/µm2.30 Despite this dense packing, rhodopsin freely diffuses in the membrane,31 which facilitates its encounter with, and activation of, transducin molecules to amplify the light signal. Many G-protein-coupled receptors are known to form dimers and higher-order oligomers. Rhodopsin has been observed to form a paracrystalline array of dimers when native disc membrane is viewed under atomic force microscopy.32 Whether it exists as dimers in vivo is still an area of controversy. However, it is known that rhodopsin is functional as monomers.33
Proteins that stabilize the structure of outer-segment discs
Rhodopsin is not only integral to the phototransduction cascade, it is also required for the formation and maintenance of OS discs; in the absence of rhodopsin, the OS structure is not formed.22 Peripherin and Rom-115,34,35 are two other disc membrane components that contribute to maintaining the flattened structure of the disc. Both of these proteins belong to the tetraspanin family of integral membrane proteins that form large multiprotein complexes known as the tetraspanin web or tetraspanin-enriched microdomains.36 Peripherin/rds is normally found within the edge or “rim” of the disc. It self-associates to form higher-order complexes and also interacts with Rom-1.37 Peripherin/rds and Rom-1 likely function as adhesion molecules that assist in keeping the discs vertically aligned, and may stabilize the disc stack with bridges to the overlying plasma membrane. Peripherin/rds is also thought to function in forming the curvature of the disc rim.38
Deletion or disruption of peripherin/rds results in malformation of discs at the OS base. The naturally occurring retinal degeneration in the mouse named “retinal degeneration, slow” (rds) has been shown to be due to a defect in peripherin/rds. The absence of peripherin/rds in the rds/rds mouse prevents normal development of the photoreceptor OS and leads to photoreceptor cell death.15,39 Transgenic mice that lack peripherin/rds fail to form an OS. When the level is reduced, as in the heterozygous mice, large whorls of disc membrane are formed instead of an organized stack of uniform discs.15,39 Mutations that affect the quaternary structure of peripherin/rds also lead to malformed discs. These observations underscore the importance of peripherin/rds in the formation and maintenance of disc structure in the OS.
Although Rom-1 is analogous to peripherin in structure and function, its absence in rods shows a less severe phenotype than peripherin/rds knockout mice, suggesting that peripherin/rds is more critical for disc formation. Although the discs grow larger than normal sizes, photoreceptor function appears to be retained to a much higher level than when peripherin/rds is absent. Nevertheless, abnormal Rom-1 does lead to slow and progressive photoreceptor degeneration both in mice and in humans.40
Functional differences of peripherin/rds and Rom-1 have been noted in rods and cones. Cones appear to have a lower ratio of Rom-1/peripherin,36 and different peripherin/rds mutations appear to affect rods and cones differentially. For example, the C214S and N244K mutations have a greater effect on rods whereas R172W and N244H tend to cause cone-dominant diseases such as macular dystrophy.36
Transmission electron microscopy has revealed structural elements between the lamellar discs as well as between the discs and the plasma membrane that are mainly localized to the rim region and incisures of discs.41–44 A study of OS structure using cryoelectron tomography of vitrified OS showed previously unobserved spacers distributed throughout the discs.30 These structural elements likely maintain the proper distance between adjacent discs and between discs and the plasma membrane. The protein identities of these spacers are not known, but peripherin/rds and Rom-1 may be candidates.
Another component of the rod disc is ABCR (photoreceptor cell-specific ATP-binding cassette transporter).45–47 Mutations in ABCR are responsible for a large variety of retinal degenerations, including Stargardt macular dystrophy, fundus flavimaculatus, some forms of cone–rod degeneration, and retinitis pigmentosa (RP). Other ABCR mutations are thought to increase the risk of developing age-related macular degeneration (AMD). ABCR is a member of the ABC superfamily. This is a large family of transmembrane proteins involved in energy-dependent transport of many different substrates across membrane “barriers.” The localization of ABCR at the disc rim suggests that it is involved in the movement of molecules from the disc lumen into the cytosol. This hypothesis was supported by studies in ABCR knockout mice, which show delayed dark adaptation, increased all-trans retinal following light exposure, and elevated phosphatidylethanolamine (PE) in the rod OS.48 Biochemical analysis of retinas from ABCR knockout mice revealed accumulation of a novel complex of all-trans retinal and PE, termed N-retinylidene-PE, which is not found in normal retina. Once in the cytosol, the all-trans retinol moves to the retinal pigment epithelium (RPE) for recycling and chromophore (11-cis retinal) regeneration (see Chapter 16, Cell biology of the retinal pigment epithelium). The primary pathologic defect in Stargardt disease, and also in ABCR knockout mice, is accumulation of toxic lipofuscin pigments, such as A2E, in RPE cells. Thus the phenotype is remarkably similar between mice and that observed in the fundus of Stargardt patients.
The animal model and further biochemical investigations49 suggest that ABCR functions as an outwardly directed flippase for N-retinylidene-PE. Delayed dark adaptation is likely due to accumulation (in discs) of the noncovalent complex between opsin and all-trans retinal. ABCR-mediated retinal degeneration in patients may result from “poisoning” of the RPE due to A2E accumulation, with secondary photoreceptor degeneration due to loss of the ABCR support role.
Though ABCR most definitely plays a role in the structure of discs, no mutations have been conclusively associated with abnormal structural features per se. In other words, it appears that mutations do not directly affect folding at the disc rim, and more likely affect the biochemical function of the transporter. In corroboration of these data, Radu and colleagues tested the effects of isotretinoin on lipofuscin accumulation in ABCR knockout mice,50 a model of recessive Stargardt disease. They observed by electron microscopy that isotretinoin blocked both the formation of A2E biochemically, and the accumulation of lipofuscin pigments. Further, no significant visual loss was observed in ABCR null mice by electroretinography (when treated), and isotretinoin also blocked the slower, age-dependent accumulation of lipofuscin in wild-type mice. The results suggest that treatment with isotretinoin may inhibit lipofuscin accumulation and delay the onset of visual loss in patients with Stargardt disease and may be an effective treatment for other forms of retinal or macular degeneration associated with lipofuscin accumulation, though “normal” visual function may be somewhat compromised by such treatment.
The presence of an OS is clearly not optional, as there are many examples of retinal disease in which the photoreceptor cell dies shortly after loss of the OS.51–53 Why loss of the OS triggers rod cell death is not obvious, since all of the required cellular organelles inhabit the IS and cell body; the OS appears devoid of organelles. The necessity to maintain an OS is further surprising since photoreceptors continually replace all the OS components. In fact, the renewal of rod cell OS components is perhaps more readily seen in the photoreceptor than in any other cell in the body. The disc and plasma membrane components of the OS are completely replaced within 2 weeks.54,55 To support this turnover there is an influx of newly synthesized proteins and lipids from the IS. These components are transported to the base of the OS, and from there discs gradually migrate towards the RPE for phagocytosis and recycling. One idea is that the high oxygen tension coming from the choroidal vasculature may interfere with the cellular machinery in the IS and nucleus of the photoreceptor, and the presence of the OS puts a protective distance between the choroid and the photoreceptor nuclei.56
Disc morphogenesis
Although it is known that new discs are formed at the base of the OS, the process by which these discs are formed is not fully resolved (Fig. 14.2). One model was described in the work of Anderson et al. (model 1).57,58 Based on morphologic studies of adult monkey rods, it was observed that the folded membrane stacks at the base of the OS are continuous with the plasma membrane of the connecting cilium, whereas the older discs are closed off and separated from the plasma membrane. From this appearance a model was proposed that the newly arrived rhodopsin-bearing vesicles fuse with the plasma membrane, and the growing membrane evaginates to form open discs. Eventually these discs pinch off and form the mature closed discs. Another model (model 2), the vesicular targeting model, was recently proposed by Chuang and colleagues.59 They provide molecular evidence that rhodopsin’s carboxyl terminus interacts with a protein called SARA (smad anchor for receptor activation), as well as phosphatidylinositol 3-phosphate (PI3P), and syntaxin 3. In their model, the new discs are already closed and enveloped by the OS plasma membrane. Their evidence suggests that the protein–protein and protein–lipid interactions organized by SARA regulate the vesicular targeting of axonemal rhodopsin-bearing vesicles to the newly formed discs. Thus the discs grow by fusing with the rhodopsin-bearing vesicles and not from the evaginated plasma membrane. This model is more consistent with the morphology of their experimental model system, the mouse rods, which do not display open discs at the base of the OS.
Outer-segment plasma membrane
It is clear that, in addition to its obvious role in phototransduction, the OS plasma membrane contains a rich array of specialized proteins, many of which regulate the movement of ions into and out of the OS. The best-characterized proteins of this type are the retinal cGMP-gated (CNG) cation channels. In the dark-adapted rods and cones, Na+ and Ca2+ flow into the OS through these channels in the plasma membrane. Calcium comprises about 10% of the dark current carried by these channels in rods,60 and perhaps 20% or more in cones.61,62 In both rods and cones Na+ is extruded from the IS through the Na+/K+ pump. This flow of ions sets up the circulating dark current, of which the vast majority is carried by the Na+. The probability of the opening of the CNG channel, which in turn determines the size of the circulating current, depends on the amount of free [cGMP], which in the dark is estimated to be 3–4 µM.63 At this concentration, the probability of channel opening is estimated to be only 0.1–0.2.64,65 This underscores the impact that elevated [cGMP] can have on the number of open channels in the diseased state. The influx of Ca2+ through the channel is balanced by an efflux of Ca2+ by the Na+/Ca2+-K+ exchanger (NCKX) in the rod OS plasma membrane, thereby maintaining the intracellular level of Ca2+ at a relatively constant level.66 Kaupp and colleagues67 cloned the cGMP channel from bovine retina, while Pittler and colleagues68 determined the primary structures of the human and mouse retinal rod cGMP-gated cation channel. These studies found that the sequence of the cGMP channel has significant similarity (59%) to the olfactory cAMP-gated channel. The retinal rod CNG channel is a hetero-oligomer composed of three alpha (CNGA1) and one beta (CNGB1) subunits,69,70 each with cytoplasmic amino (N)- and carboxyl (C)-termini, six putative transmembrane domains, and a pore region.71,72 Mutations in CNGA1 and CNGB1 have been linked to disease. A point mutation in CNGB173 and several mutations in CNGA174 have been identified and linked to recessive RP in humans. The CNG channel in cones consists of two CNGA3 and two CNGB3 subunits.75 Together, mutations in either of these genes account for ~75% of complete acromatopsia.76,77
In addition to the CNG channel, there are several other channels and transporters in the plasma membrane that serve to regulate the intracellular contents of the OS. The best studied of these is the NCKX. Rods express NCKX1 whereas cones express NCKX2.78 The exchanger moves with every cycle 4 Na+ into the OS, and in exchange, moves one Ca2+ and one K+ into the subretinal space; exchange is thus electrogenic. Other transmembrane proteins, such as the GLUT-1 glucose transporter, have been shown to be present on both rod and cone OS.79
Outer-segment lipids
Docosahexaenoic acid (DHA, 22:6n-6) is the major fatty acid found in the retinal rod OS, and rod photoreceptors have higher levels of DHA than in any other membrane system examined.80 Numerous studies have established that the high level of DHA in rod OS membranes provides an optimal microenvironment for rhodopsin. DHA belongs to the n-3 family of essential polyunsaturated fatty acids. These fatty acids cannot be synthesized by vertebrates and they, or their shorter-chain precursors, must therefore be obtained in the diet.81
Humans with RP and dogs with progressive rod–cone degeneration (prcd) have lower than normal blood levels of long-chain polyunsaturated fatty acids, including DHA. In addition, prcd-affected dogs have lower levels of DHA in their rod OS than control animals.82 The reason for the reduced level of DHA in rod OS of animals with inherited retinal degeneration is not known. However, the fatty acid composition of the rod OS of these animals suggests that the synthesis of DHA-containing glycerolipids is downregulated in retinas of animals with inherited retinal degenerations.
Phototransduction
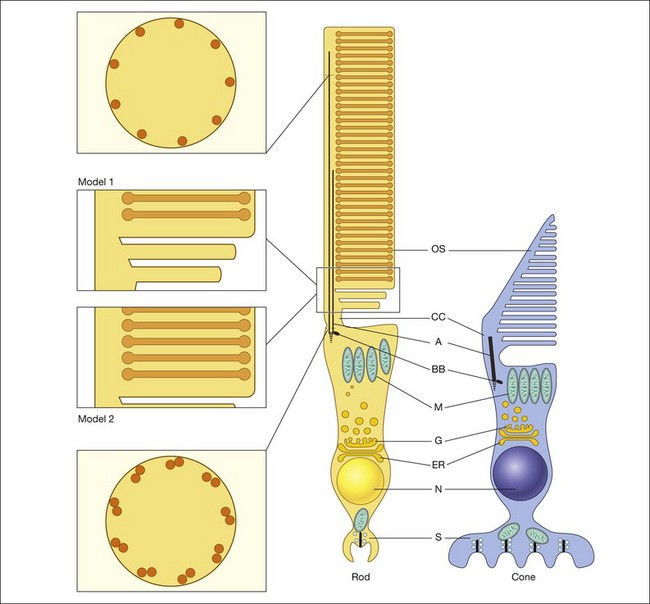
Visual transduction is initiated by the phototransduction cascade, which is perhaps the best characterized of all G-protein receptor-coupled signaling pathways. Our knowledge about phototransduction is relatively advanced because the components of this pathway are located selectively in the photoreceptor OS and can be isolated in quantities suitable for biochemical analysis. Photoreceptors can also be isolated easily for electrophysiological measurements, either as single cells using suction electrodes (Fig. 14.3), or en masse using field potentials called electoretinograms (ERG: see Chapter 7, Electrogenesis of the electroretinogram). The phototransduction cascade within rods is so robust that single-photon absorption by rhodopsin gives rise to a change in current that can be monitored by suction electrode recordings. Similarly, the remarkable ability of cone phototransduction to adapt to increases in background light intensity can also be monitored with suction electrodes. The combination of transgenic technology and electrophysiological analysis has greatly enhanced our understanding of signal transduction within these photoreceptor cells. In particular, transgenic technologies have allowed the introduction of targeted changes into specific components of the signaling pathway. These genetically altered intact photoreceptor cells can then be subjected to electrophysiological measurements, and their tissues can subsequently be used in biochemical or immunocytochemical assays to pinpoint the molecular mechanism behind the physiological phenotype. In this section we summarize the current state of our understanding of phototransduction in retinal photoreceptors.
Signal activation and amplification
A hallmark of rod phototransduction is its high sensitivity. Fully dark-adapted rods achieve sensitivity that reaches the theoretical maximum, the ability to detect individual quanta of light.1,2 This high sensitivity is generated through several stages of amplification, resulting in a tremendous increase in the signal gain (Fig. 14.4). This cascade of amplification begins at the light-activated G-protein-coupled receptor rhodopsin (R). Absorption of a photon by R leads to a conformation change (R*), allowing it to interact with the heterotrimeric G-protein transducin (Tα, β, and γ), thereby promoting the exchange of bound GDP for GTP. The complex then dissociates into Tα-GTP and Tβγ, and R* is free to activate many other transducin molecules during its catalytic lifetime.83 In the subsequent step, Tα-GTP binds and removes the inhibitory γ-subunit of the cGMP-PDE so that it can now hydrolyze cGMP to 5’-GMP; the enzymatic activity of PDE (PDE*) in turn degrades thousands of cGMP molecules. The decrease in cytoplasmic cGMP concentration then leads to closure of the cGMP-gated cation conductance channel in the rod plasma membrane, resulting in a reduction in the influx of ~ 1 000 000 Na+ ions.84 This is a graded effect: a slight lowering in the cGMP concentration leads to closure of some channels, while a large decrease will eventually lead to closure of all channels. In this instance, the rod cell is said to be saturated.
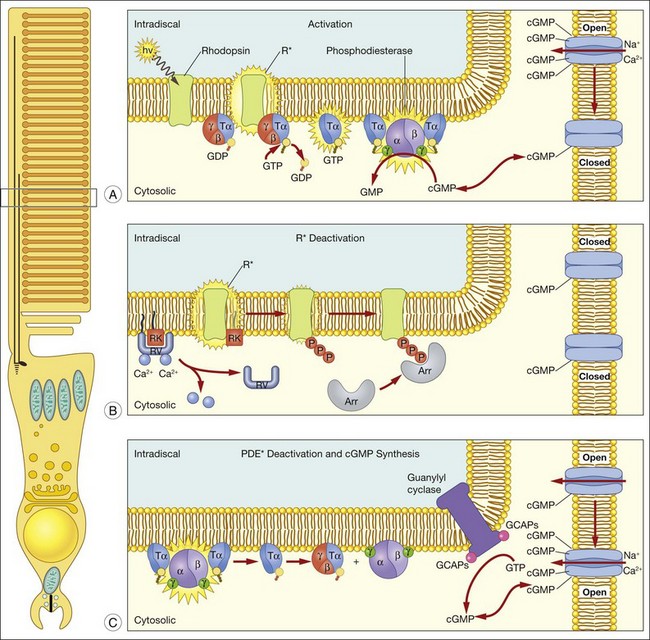
In darkness, glutamate, the transmitter used by both rods and cones, is released at a steady rate because the photoreceptor’s membrane potential is in a relatively depolarized state. The closure of CNG channels results in a graded hyperpolarization of the cell, leading to a decrease in glutamate release at the synapse. In this manner, the first sensation of light perception is transmitted from the photoreceptor cell to second-order cells of the retina, where signals are further processed and ultimately conveyed to the retinal ganglion cells (see Chapter 15, Functional anatomy of the mammalian retina).
Signal deactivation
A reversal of the activation steps is required ultimately for the photoreceptor to return to its resting state. First, the catalytically active components of the phototransduction cascade, R* and PDE*, must be quenched, then cGMP needs to be resynthesized (Fig. 14.4). These events must be rapid and reproducible for the photoreceptor to maintain its high sensitivity and respond to subsequent stimuli. Our current understanding of these processes is detailed below.
Quenching R*: phosphorylation and arrestin binding
Since the early 1980s, phosphorylation was recognized to play an important role in deactivation of R*.85,86 To determine how receptor deactivation occurs in vivo, transgenic mouse models were developed to assess the contribution of receptor phosphorylation to R* shutoff. In early experiments, a rhodopsin truncation mutation, S334ter, was expressed in the photoreceptors of transgenic mice17 that removed the terminal 15 amino acid residues and thus all putative Ser and Thr phosphorylation sites. Single-photon responses produced by S334ter R* failed to shut off in a timely and stereotyped manner, indicating that this domain is important for R* quenching. In the 1990s, numerous biochemical experiments suggested that specific Ser residues on the C-terminus are crucial in R* shutoff.87–89 However, single-cell recordings from transgenic mice rods expressing rhodopsin where these phosphorylation sites removed in different combinations showed that all of the Ser and Thr sites at the C-terminus are required for rapid and reproducible R* deactivation; the cluster of Ser and Thr sites at rhodopsin’s C-terminus functions to ensure fast and reproducible R* deactivation.90 It is clear that rhodopsin kinase is solely responsible for phosphorylating R*, since phosphorylation of R* does not occur in mice lacking rhodopsin kinase.91
Subsequent to C-terminus phosphorylation, the catalytic activity of R* is quenched fully by binding of visual arrestin. Arrestins are soluble cytoplasmic proteins that bind to G-protein-coupled receptors, thus switching off activation of the G-protein and terminating the signaling pathway that triggers the cellular response; the most commonly studied arrestin is β-arrestin. Visual arrestin exhibits exquisite specificity to phosphorylated R*. Our understanding of how this specificity is conferred emerges from extensive mutagenesis studies, and the crystal structure of visual arrestin.92–95 These studies reveal that arrestin is constrained into a latent, inactive structure by a network of intramolecular interactions.92 According to the current model of arrestin activation, these intramolecular constraints are released by: (1) interaction with multiple phosphates on rhodopsin’s C-terminus; and (2) interaction with the cytoplasmic loops of R*. The physiological role of visual arrestin in controlling the rod single-photon response was studied by Xu and colleagues,24 who created transgenic mice lacking visual arrestin. Suction electrode recordings of photoresponses from the OS of these transgenic rods displayed a rapid, partial recovery, followed by a prolonged final recovery phase. The timing of the slow phase of shutoff is consistent with the spontaneous decay of the R*. Thus, while R* phosphorylation is required to terminate rapidly its catalytic activity, the full quenching of R* activity requires visual arrestin binding.
Deactivating PDE: control of transducin’s GTPase activity
Quenching of the phototransduction cascade also requires the shutoff of PDE*, which occurs through the deactivation of Tα. The GTPase activity of Tα allows the reassociation of the inhibitory γ-subunit to the catalytic PDE subunits,96 a process which is speeded through the participation of a photoreceptor-specific RGS protein, RGS-9, and its binding partner, Gβ5.97 These proteins act sequentially on Tα to facilitate the hydrolysis of bound GTP. The physiological role of all these proteins in the shutoff of the rod photoresponse was evaluated with transgenic mice that had alterations in each of these components. Rod photoresponses from transgenic mice expressing a PDE-γ with an impaired ability to stimulate GTP hydrolysis98 showed abnormally slowed recovery, demonstrating the necessary role of PDE-γ in Tα deactivation.99 Similarly, mice lacking RGS-9 showed profoundly slowed response recovery,100 as do mice lacking Gβ5.101 RGS-9 is anchored to photoreceptor membranes by the adaptor protein, R9AP.102–104 Interestingly, human patients with mutations in R9AP have difficulties adapting to sudden changes in light levels that are mediated by cones,105 consistent with the higher concentration of RGS-9 complex in cones compared to rods.106 Together, the current data support an important role of PDE-γ, RGS-9/Gβ5, and R9AP in Tα deactivation, and consequently, response recovery, in both rods and cones.
Resynthesis of cGMP: Ca2+ dependence of guanylyl cyclase
Recovery of the dark current also requires that cGMP is resynthesized to allow CNG channels to open. cGMP is synthesized in photoreceptor OS by a retinal guanylyl cyclase (RetGC), which is regulated by the protein GCAPs (GCAP 1 and 2).107 GCAPs is an EF hand Ca2+-binding protein that confers on RetGC a highly cooperative Ca2+ dependence for cGMP synthesis.108 Near the dark resting Ca2+ concentration RetGC’s activity is inhibited, but as OS Ca2+ falls in response to illumination as CNG channels close, RetGC’s activity accelerates to restore the resting cGMP and reopen CNG channels. The physiological actions of GCAPs have been evaluated in physiological recordings. For instance, when GCAP1 is dialyzed into gecko rods, the recovery phase of the light response is accelerated.109 In addition, suction electrode recordings from rods of GCAPs knockout mice show that the Ca2+ dependence of cGMP synthesis is required to set the time course of the rod’s photoresponse, and to suppress noise within the phototransduction cascade.110
Light adaptation
In a normal cycle of day and night, the illumination at the Earth’s surface varies over 11 orders of magnitude, making photoreceptor adaptation fundamentally important to the normal functioning of the vertebrate visual system. Rods cells are able to adjust their sensitivity over 2–3 log units of light intensities, while cone cells exhibit no response saturation over 6–7 log units.3,4 The switch between rod and cone function from night to bright daylight covers a large portion of vision’s functional range, and downstream circuitry accounts for the remainder of the range extension. While the molecular events underlying the amplification cascade following photon capture by rhodopsin are well delineated, the mechanisms underlying photoreceptor adaptation are less well understood; however, Ca2+ is known to play a feedback role in the adaptation process.111,112
The role of Ca2+ feedback
In darkness, Na+ ions enter the OS through the CNG channels and are extruded in the IS through Na+/K+ ATPase pumps. This steady-state influx and efflux of Na+ is largely the basis for the circulating dark current in photoreceptor cells. Similarly, Ca2+ enters through CNG channels in the OS, but is extruded locally by NCKX exchangers. In mouse rods the steady-state influx and efflux of Ca2+ hold concentration near 250 nM,113 but as light closes CNG channels, Ca2+ entry is blocked, Ca2+ efflux continues, and [Ca2+]i falls. This change in [Ca2+]i is graded just like the dark current: the degree of lowered [Ca2+]i is proportional to the intensity of background light.
This decrease in [Ca2+]i triggers a feedback signal that is necessary for light adaptation, since, when the [Ca2+]i decrease is blocked, adaptation to background illumination is greatly compromised.114,115 Existing evidence suggests that this feedback is rather complex, as it orchestrates several pathways directed toward different components of the transduction machinery.116–118 Here we review the role of Ca2+ on each of these components.
Adaptation mediated by Ca2+ feedback to retinal guanylyl cyclase
It has long been recognized that feedback controlling accelerated recovery contributes to light adaptation.108 Recovery of the light response requires the resynthesis of cGMP, which is needed to reopen the CNG channels and re-establish the circulating current. The control of cGMP synthesis by RetGC/GCAPs is central to this process. The contribution of accelerated cGMP synthesis to adaptation in rod photoreceptors was investigated in GCAP knockout mice.23,110 This work showed that the presence of GCAPs extends the sensitivity to background light in rod photoreceptors ~10-fold (Fig. 14.5), and can account for about half of the rod’s capacity to adapt to background light. Recordings from cones in GCAPs knockout mice also show that GCAP contributes to background adaptation, but to a lesser extent than it does in mouse rods.119
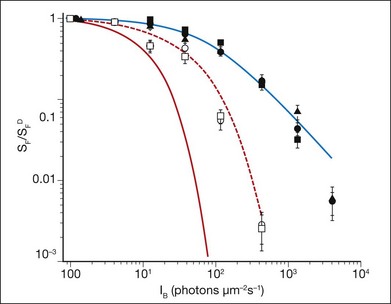
Fig. 14.5 Sensitivity as a function of background light was measured from mouse rod photoreceptors using suction electrodes (Fig. 14.3). Sensitivity was calculated from dim flash responses as the peak response amplitude divided by the flash strength. Flash sensitivity (SF) in the presence of increasing background light, background light (IB) was normalized against the flash sensitivity in darkness (SFD) and is plotted for the rods of several transgenic mice with particular elements of the phototransduction cascade altered. These mice include wild-type (•), mice lacking recoverin ( ), mice where the calmodulin-binding site on the cyclic nucleotide gated (CNG) channels has been eliminated (
), mice where the calmodulin-binding site on the cyclic nucleotide gated (CNG) channels has been eliminated ( ), mice lacking guanylyl cyclase-activating proteins 1 and 2 (
), mice lacking guanylyl cyclase-activating proteins 1 and 2 ( ), and mice where both the calmodulin-binding site on the CNG channel and guanylyl cyclase-activating proteins 1 and 2 have been eliminated (
), and mice where both the calmodulin-binding site on the CNG channel and guanylyl cyclase-activating proteins 1 and 2 have been eliminated ( ). The red line shows the predicted decline in sensitivity as a function of background light intensity if rods displayed no light adaptation. The blue line is best-fitting Weber–Fechner function for wild-type rods, and shows that light adaptation extends the sensitivity of rod photoreceptors to 100-fold higher background light levels. Rods from mice lacking recoverin, or the calmodulin-binding site on the CNG channels, show little deviation from wild-type rods, indicating that these proteins do not play a significant role in light adaptation. However, in mice lacking guanylyl cyclase-activating protein 1 and 2, light adaptation is impaired, but not absent, even when the calmodulin-binding site on the CNG channel is also eliminated (red dashed line). The remaining mechanisms that control light adaptation in rod photoreceptors remain unidentified.
). The red line shows the predicted decline in sensitivity as a function of background light intensity if rods displayed no light adaptation. The blue line is best-fitting Weber–Fechner function for wild-type rods, and shows that light adaptation extends the sensitivity of rod photoreceptors to 100-fold higher background light levels. Rods from mice lacking recoverin, or the calmodulin-binding site on the CNG channels, show little deviation from wild-type rods, indicating that these proteins do not play a significant role in light adaptation. However, in mice lacking guanylyl cyclase-activating protein 1 and 2, light adaptation is impaired, but not absent, even when the calmodulin-binding site on the CNG channel is also eliminated (red dashed line). The remaining mechanisms that control light adaptation in rod photoreceptors remain unidentified.
(Data replotted from Chen J, Woodruff ML, Wang T, et al. Channel modulation and the mechanism of light adaptation in mouse rods. J Neurosci 2010;30:16232–40.)
Recoverin and control of rhodopsin kinase
The accelerated response recovery that mediates light adaptation is also achieved through the speeded deactivation of R*. The first step in the quenching of R* activity is the phosphorylation of its C-terminus by rhodopsin kinase, a process that is believed to be regulated in a Ca2+-dependent manner by the protein recoverin. Recoverin is a highly conserved Ca2+-binding protein found in both rod and cone photoreceptors. In vitro, recoverin binds and inhibits rhodopsin kinase when it is Ca2+-bound, and accelerates rhodopsin kinase activity as Ca2+ is removed.120,121 Since [Ca2+]i is high in darkness, it has been suggested that, under dark-adapted conditions, recoverin prolongs the catalytic activity of R* by inhibiting rhodopsin phosphorylation. Early evidence in experiments that introduce recombinant recoverin into gecko and salamander rods indicated that recoverin prolongs the light response.122,123 However, recoverin’s effect was most prominent at Ca2+ concentrations outside the rod’s physiological range (>1 µM). The Ca2+ requirement for rhodopsin kinase inhibition by recoverin in reconstituted biochemical assays was similarly high.120 These results placed into doubt the role of recoverin in mediating light adaptation. Suction electrode recordings from the rods of transgenic mice lacking recoverin, however, reveal a response speeding.124 This speeding effect is exacerbated in rods that lack recoverin, but also have a reduced expression of rhodopsin kinase.125 Thus, in the physiological range of Ca2+ concentrations, recoverin can influence R* lifetime. Despite this effect on R* lifetime, recoverin appears to play a relatively minor role in rod light adaptation (Fig. 14.5).124 It remains to be seen in cone photoreceptors what is the efficacy of recoverin’s action. However, one might expect a greater contribution to adaptation as experiments from salamander L cones show that R* lifetime dominates the light response.126
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


