Chapter 130 Capillary Hemangioblastoma of the Retina and von Hippel–Lindau Disease
Von hippel–lindau disease
Von Hippel–Lindau (VHL) (MIM193300) is an autosomal dominant neoplastic disorder in which multiple benign or malignant tumors and cysts with specific histopathologic features develop in the tissues of the central nervous system: the brain, spinal cord, inner ear, and retina and in visceral organs: the kidney, adrenal gland, pancreas, epididymis and broad ligament.1 VHL is a rare disorder (with an approximate incidence of 1 in 36 000 live births) and has a penetrance of over 90% by 65 years of age.
History
Over a century ago, von Hippel described retinal capillary hemangioblastomas transmitted through several generations of family members in a small number of pedigrees.2 In 1926, the Swedish pathologist, Lindau, observed the link between retinal and cerebellar hemangioblastomas as well as cysts in the kidney, pancreas, and epididymis as part of a familial syndrome.3 Melmon and Rosen established the first clinical diagnostic criteria for VHL in their landmark summary manuscript in 1965.4 Seizinger and colleagues discovered the linkage of the VHL gene to the short arm of chromosome 3 in 1988.5 Latif and colleagues identified the VHL tumor suppressor gene in 1993.6 The disease results from a germ line mutation in the VHL gene, which is located on the short arm of chromosome 3, 3p25–26.6
Genetic associations
In 1993, the discovery of the VHL gene,6 located on the short arm of chromosome 3 (3p25–26), was a seminal step in the molecular study of VHL disease. The isolation of the gene for VHL disease has led to significant advances in understanding how the VHL protein functions and how its inactivation may result in disease. The VHL gene is ubiquitously expressed and the protein acts to degrade particular transcription factors called hypoxia inducible factors (HIFs).7 HIFs are produced in response to low tissue oxygen levels, or hypoxia, and serve to upregulate proteins that can help reverse hypoxia, such as erythropoietin (EPO), vascular endothelial growth factor (VEGF), and platelet-derived growth factor (PDGF-β).8 Loss of VHL function by mutation of the VHL gene results in unregulated high levels of HIF, which in turn produces high levels of downstream gene products including VEGF, erythropoietin, PDGF-β, and transforming growth factor-α (TGF-α).9–11 The vascular nature of the VHL tumors may result from the HIF-mediated increased levels of VEGF and PDGF-β, which are known to support the proliferation of endothelial cells and pericytes, respectively.12 Moreover, increased vascular permeability of the tumor vessels resulting from increased levels of VEGF may also account for the edema seen with lesions of VHL.
The mutations in the VHL gene that result in VHL disease are highly varied, and may range from single base pair substitution in a single amino acid codon to the complete deletion of the gene.13,14 Although many of these mutations are thought to impair the ability of VHL to regulate HIF, the diversity in the manifestations of VHL disease suggests that different mutations may actually affect VHL protein function in different ways, and possibly even confer novel functions.15 Correlating the genotype to the phenotype might help elucidate how different types of VHL mutations result in disease. For example, the location of the missense mutation in the VHL gene correlates significantly with the prevalence and the phenotype of the eye disease, which would also in turn influence visual acuity loss in the affected patients.16 Such analyses might also help us understand the mechanisms by which the VHL mutations affect the eye.
Clinical presentation
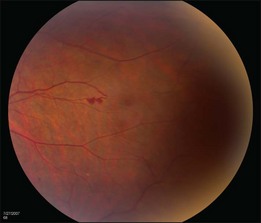
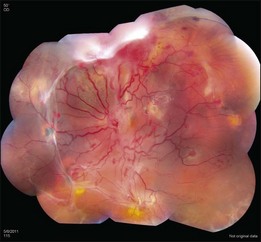
The retinal capillary hemangioblastomas, often the first manifestation of VHL, range from very small capillary abnormalities (Fig. 130.1) to large lesions that cause major visual impairment. The clinical appearance of the retinal capillary hemangioblastoma is very typical and diagnostic of VHL. The initial appearance of a retinal capillary hemangioblastoma is a subtle red or grayish dot no larger than a few hundred microns. As the proliferation of the vascular tumors (mostly composed of capillaries, with often a fibrotic component) progresses, secondary alterations occur to produce a distinctive clinical appearance that is often nodular (Fig. 130.2). The blood vessels leading to and away from the tumor become characteristically dilated and markedly enlarged (Fig. 130.3). This tumor, although often located in the retinal periphery, can lead to edema and hard exudates both around the tumor and in the center of the macula (Fig. 130.4). Without treatment, retinal capillary hemangioblastomas can grow and displace the normal structures of the retina and also cause an exudative retinal detachment. The fibrosis that accompanies these hemangioblastomas may result in tractional retinal detachment, with hemorrhaging and finally, neovascular glaucoma. Rarely, these tumors regress spontaneously.17 The tumors are located predominantly in the retinal periphery and less frequently on or around the optic disc (see below).
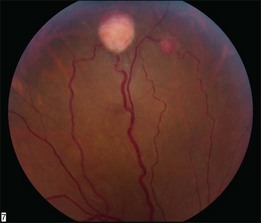
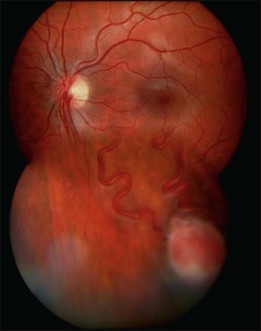
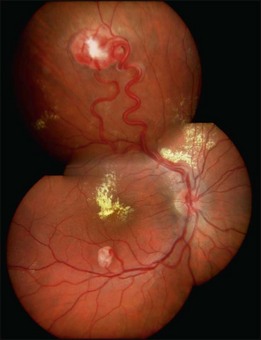
Optic nerve capillary hemangioblastomas or juxtapapillary capillary hemangioblastomas can start as small lesions at the optic disc or in the peripapillary area (Fig. 130.5). They can remain relatively slow growing for many years. However, with either increased growth (Figs. 130.6) or increased vasopermeability, exudative changes can occur (Fig. 130.7). Retinal edema, with retinal hard exudates, especially localized to the macula, can decrease vision. In addition, these exudative changes can lead to exudative retinal detachment (Fig. 130.8).
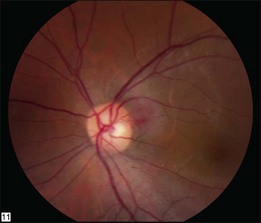
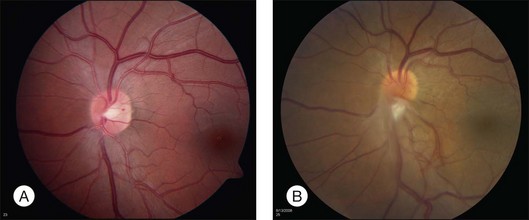
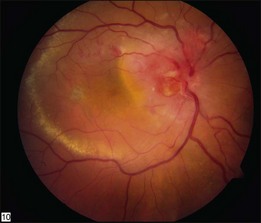
Fig. 130.7 Optic nerve tumor of this right eye has resulted in macular edema and marked retinal hard exudate.
Patients are often asymptomatic and VHL lesions tend to progress slowly. The retinal lesions can be detected in children to adults in the eighth decade of life on a routine exam.18,19 Symptoms such as decreased vision or strabismus may result in the detection of VHL in children. Decreased visual acuity can also cause adults to seek medical help with subsequent detection of the disease. The ophthalmologist plays an important role in initiating the medical work-up for VHL. Genetic testing has proven to be helpful in early diagnosis and clinical screening for VHL.
Clinical diagnosis
The diagnosis of the retinal capillary hemangioblastoma is primarily clinical, while other studies may help to confirm the diagnosis; there is no pathognomonic diagnostic tool. Fundus photography, especially with montage techniques or a wide-angle camera to capture the location and size of the peripheral lesions may be helpful in following the growth or regression of the lesions. Fluorescein angiography typically shows early leakage and marked hyperfluorescence and this may persist or decrease in the late phases of the study. Macular edema associated with these lesions may also be detected on fluorescein angiography. Optical coherence tomography may also be useful in such cases of macular edema but adds little to the diagnosis of retinal capillary hemangioblastoma (Fig. 130.9). Ultrasonography shows an acoustically solid retinal mass with a smooth anterior border with variable but mostly medium reflectivity (Fig. 130.9). Orbital shadowing and choroidal excavation are not seen.
Epidemiology of ocular lesions of von hippel–lindau disease
The true prevalence of the ocular manifestations of this rare systemic disease is difficult to ascertain. Most case series have reported higher prevalence of ocular disease as patients are referred to the ophthalmologists because of visual symptoms.20 The prevalence of ocular VHL was assessed in a study in which patients were referred because they were identified in the community as affected with VHL and not because of visual symptoms.21 The affected probands and their family members were invited to participate in a study at the National Institutes of Health for screening of VHL and its complications. This series of patients was diagnosed, examined and treated for various lesions of VHL by a team of experts including genetic counselors, medical geneticists, radiologists, pathologists, neurosurgeons, urologists, radiation oncologists, ophthalmologists, and otolaryngologists.
Over the period of 1988–2000, 335 (38%) affected individuals from a cohort of 890 patients of 220 unrelated pedigrees were found to have ocular involvement.21 Of those affected with ocular VHL, 42.1% were unilateral and 57.9% were bilateral. Approximately 85% were located in the peripheral retina, while 15% were juxtapapillary. Their ages ranged from 7 to 84 (mean 36 years with standard deviation ± 15 years), and 189 (47%) were male. In this population, 386 (95%) were white, seven (1.7%) were black, ten (2.5%) were Hispanic, and three (0.7%) were Asian.
Causes of vision loss
Visual loss from RCHs is generally caused by exudation from the tumor, causing retinal edema, or glial proliferation on the surface of the tumor resulting in retinal striae and distortion and eventually tractional and/or exudative retinal detachment.10 Interestingly, while tumor numbers did not increase significantly as a function of age, the risk of vision loss was found to increase with age, as was found in another cross-sectional series.22 The risk of severe vision loss also increased with the presence of juxtapapillary lesions and increasing peripheral tumor number and the extent of the retinal involvement.11 In the cohort studied at the National Eye Institute, approximately 77% had vision of 20/20 or better. The overall prevalence of legal blindness from ocular VHL was low at 5.7% with vision less than 20/160 in the better-seeing eye. Although this number appeared low, about 20% of all the patients with ocular VHL had some unilateral visual impairment, thus the burden of disease is still moderately high.
In about 8% of the NEI population, patients were found to have retinal neovascularization that mimicked diabetic retinal neovascularization.23 The retinal neovascularization most likely represents one end of the spectrum of hemangioblastoma formation. These patients tended to be younger and a majority of them had it as the sole finding in one eye, while the other eye had evidence of retinal capillary hemangioblastoma. The retinal neovascularization was also found often at the optic disc with an epiretinal membrane. The removal of the neovascularization along with the epiretinal membrane with vitrectomy and membrane peeling can result in visual acuity improvement. Other retinal lesions seen on the optic nerve include retinal vascular hamartomas.
Patients with systemic VHL also experienced vision loss from neurological lesions such as expanding cerebellar hemangioblastomas that may produce sustained elevated intracranial pressure, causing pressure on the optic nerves and resultant optic atrophy. Following surgical resection of the cerebellar lesions, the visual acuity can usually be expected to improve; but rarely, visual impairment may remain. Other extraocular causes of visual loss include hemangioblastomas that occur in the retrobulbar space, intracranial optic nerve, chiasm and/or the optic tract.24–29 Approximately 5.3% of cases of intracranial optic nerve/tract hemangioblastomas were documented in the NEI population.
Other manifestations of VHL that may come to an ophthalmologist’s attention include hypertensive retinopathy from pheochromocytomas, retinal vascular hamartomas,30 chiasmal syndromes,31 afferent pupillary defects from optic nerve hemangioblastomas,28 and papilledema (due to CNS lesions, optic nerve lesions, or pseudopapilledema with peripapillary RCH).32–34 Retinal function in general may be affected in VHL patients as demonstrated by electroretinography, although the mechanism for this dysfunction is obscure.35
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


