Chapter 80 Rheumatic Disease
An approach to the assessment of the patient with possible rheumatic disease
Common ocular presentations of rheumatic disease
Keratoconjunctivitis sicca and other corneal presentations
Keratoconjunctivitis sicca (KCS, or “dry eye syndrome”) is a common ocular manifestation of a number of rheumatic diseases including rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), scleroderma, and relapsing polychondritis. Symptoms vary from slight irritation and burning in mild disease to severe pain and blurred vision arising from increasing corneal involvement. Clinical examination using slit-lamp biomicroscopy reveals a small or absent tear meniscus with a tear film break-up time of less than 10 seconds. Corneal abnormalities, which may be highlighted with fluorescein drops and a cobalt blue light, include punctate epitheliopathy, mucus filaments, strands, and plaques. Additional staining with Rose Bengal or Lissamine Green drops reveals a characteristic interpalpebral pattern, with greatest staining nasal and temporal to the corneal limbus. Tear production, as measured by Schirmer’s test, is reduced. Wetting of the test strip by less than 5 mm after 5 minutes in the un-anesthetized eye indicates severe tear deficiency. It should be noted that the correlation of dry eye symptoms with observed disease is poor. Many more patients report “dry eyes” than have visible disease, and many asymptomatic patients do have some degree of keratoconjunctivitis sicca.1
Other less common corneal presentations of rheumatic disease include the sight-threatening peripheral ulcerative keratitis (PUK). The etiology is uncertain but it has been suggested that immune complex deposition at the corneal limbus results in an obliterative vasculitis and stromal melt. It is most commonly associated with RA or systemic vasculitis, in particular granulomatosis with polyangiitis (previously known as Wegener’s granulomatosis).2,3 Clinical features include variable pain and redness and reduced vision, uni/bilateral peripheral corneal ulceration with epithelial defect and stromal thinning, associated limbal inflammation, and scleritis.
Scleritis/episcleritis
Scleritis
Scleritis is associated with systemic disease in around 40–50% of patients, of which most are cases of a rheumatic disease, such as RA, granulomatosis with polyangiitis, relapsing polychondritis, SLE, sarcoidosis, polyarteritis nodosa, inflammatory bowel disease, psoriatic arthritis, ankylosing spondylitis, and gout.4 It is commonest in middle-aged women. Scleritis is bilateral in 50% of cases, but both eyes may not be affected at the same time.5 The pain (constant/deep/boring) can be so severe that it may wake the patient at night. The eye has an intense red/dark red appearance. The globe may be very tender to touch. A bluish hue implies scleral thinning from previous active scleritis due to the underlying blue/black uveal tissue showing through the translucent sclera. Scleral thinning can eventually result in high degrees of astigmatism. The degree of redness and scleral thinning is more easily seen under room light or in daylight than by the slit lamp. Topical phenylephrine 2.5% causes blanching of the more superficial episcleral vessels but does not change the engorgement of deeper scleral vessels and can often help differentiate between scleritis and episcleritis. The most severe type is necrotizing anterior scleritis with inflammation. Apart from the severe pain and redness, there may be tearing and photophobia. White avascular areas surrounded by injected edematous sclera are present that may lead to scleral necrosis. An associated anterior uveitis suggests advanced disease. Complications of scleritis include peripheral ulcerative keratitis, acute stromal keratitis, sclerosing keratitis, uveitis, cataract, astigmatism, glaucoma, and globe perforation.
Episcleritis
This common condition is a benign, recurrent inflammation of the episclera. Being superficial it is easily distinguished from deeper scleral inflammation, in that it is less painful and the involved vessels blanch on instillation of topical phenylephrine 2.5%. It is more common in young women, often self-limiting, and may require little or no treatment. It is not usually associated with any systemic disease, although around 10% may have an underlying rheumatic disease.6
Uveitis
Acute anterior uveitis
In acute anterior uveitis (AAU) patients typically present with pain, photophobia, redness, and blurred vision. Examination findings are of anterior segment inflammation including circumlimbal injection, keratic precipitates (especially inferior), anterior chamber (AC) flare, cells, and fibrin (fibrin is a key feature in HLA-B27-associated uveitis). A hypopyon is suggestive of HLA-B27-associated disease, Behçet disease, or severe intraocular infection.7 Posterior synechiae are common in both idiopathic and HLA-B27-associated AAU and every effort should be made to break them at time of presentation. Vitreous cells may be seen as “spill-over” inflammation, but vitritis is not a dominant feature. Occasionally cystoid macular edema (CME) may be seen (especially in HLA-B27 disease), but this is more commonly a feature of intermediate, posterior or pan-uveitis. It is estimated that up to one-third of patients with AAU have ankylosing spondylitis (AS).8 Treatment is with intensive topical corticosteroid and mydriatic. If severe, subconjunctival corticosteroid and mydriatic, oral corticosteroid or even intravenous corticosteroid may be given. Recurrent disease (especially if frequent or severe) may be an indication for maintenance systemic treatment.
Therapeutic considerations
Many patients will require systemic treatment, such as corticosteroids, immunosuppressants (methotrexate, azathioprine, mycophenolate mofetil, cyclosporine), and biologics (anti-TNF, rituximab – anti-CD20) for their rheumatic and ophthalmic disease. Where possible this should be undertaken in conjunction with a rheumatologist who has an expertise in this type of therapy. Ophthalmologists should only prescribe these drugs if they have a detailed knowledge of their action, route of administration, side-effects, and how to monitor for them, as the treatments themselves have potentially serious complications. Most patients with sight-threatening disease are likely to be prescribed oral corticosteroid, and these have numerous, well-recognized side-effects, including glucocorticoid-induced bone disease.9 Fortunately, in patients with ocular inflammation the commonly used immunosuppressants do not appear to increase overall or cancer mortality.10
Disease-specific section
Rheumatoid arthritis
Epidemiology
RA is the commonest of the inflammatory arthritides, with an incidence of around 3 in 10 000 per annum and a prevalence of 1% among adults in industrialized nations.11–13 Epidemiological risk factors include age (increases with age), female gender (3–5 times greater risk than male),13 and smoking.14
Articular and systemic disease
Typical features of the arthritis of RA are its symmetrical small joint distribution and its deforming nature, giving rise to the classic appearance of ulnar deviation (“rheumatoid hands”). All synovial joints may be affected, including the metacarpophalangeal joints (MCP), proximal interphalangeal joints (PIP), the interphalangeal joint of the thumb, metatarsophalangeal joints, wrist, elbows, hip joints, knees, and the atlanto-axial joint.15 Degenerative changes of the atlanto-axial joint may make intubation hazardous and should be considered when planning anesthesia. In contrast to noninflammatory degenerative arthritis such as osteoarthritis, patients commonly complain of morning stiffness that improves with exercise.15
Extra-articular features are common and affect many systems.16 One-quarter of patients with RA develop solid lesions within the subcutaneous tissues of extensor surfaces known as rheumatoid nodules. Cardiac complications includes accelerated atherosclerotic coronary artery disease, pericarditis, heart block or valvular dysfunction.17 Respiratory complications include pleural effusions, nodules of the pleura or lung tissue, and interstitial fibrosis; a severe variant of interstitial fibrosis associated with RA and coal-miners’ pneumoconiosis is known as Caplan syndrome.18 Other systemic complications include renal amyloidosis and a chronic anemia and/or leucopenia. The combination of RA, splenomegaly, and leucopenia is known as Felty syndrome.19 Vasculitis is a rare but important complication of RA which ranges in severity from nail-fold infarcts to severe life-threatening systemic vasculitis.20
In addition to clinical assessment, the diagnosis may be supported by systemic investigations such as the measurement of inflammatory markers, rheumatoid factor, and anticitrullinated protein antibodies (ACPA) (Box 80.1).21
Box 80.1
The 2010 American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) classification criteria for rheumatoid arthritis21
Classification criteria for RA
| Score | |
| A. Joint involvement | |
| 1 large joint | 0 |
| 2–10 large joints | 1 |
| 1–3 small joints (with or without involvement of large joints) | 2 |
| 4–10 small joints (with or without involvement of large joints) | 3 |
| >10 joints (at least 1 small joint) | 5 |
| B. Serology (at least 1 test result is needed for classification) | |
| Negative RF and negative ACPA | 0 |
| Low-positive RF or low-positive ACPA | 2 |
| High-positive RF or high-positive ACPA | 3 |
| C. Acute-phase reactants (at least 1 test result is needed for classification) | |
| Normal CRP and normal ESR | 0 |
| Abnormal CRP or abnormal ESR | 1 |
| D. Duration of symptoms | |
| <6 weeks | 0 |
| ≥6 weeks | 1 |
(Adapted from Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid Arthritis Classification Criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569–81.)
Ocular disease
Scleritis occurs in 1–6% of patients with RA, and in up to 14% of patients with rheumatoid vasculitis.2,4,5 Scleritis in the context of RA may present with severe pain and may be diffuse or nodular, anterior or posterior, and necrotizing or non-necrotizing in pattern.6 Of greatest concern is scleromalacia perforans, in which scleral destruction is not typically painful and not associated with visible signs of inflammation.2,6 Episcleritis may also be seen in the context of RA.6,22
Corneal complications of RA are wide-ranging with varying risk of perforation. Marginal keratitis without apparent inflammation may result in peripheral thinning giving rise to the appearance of “contact lens cornea”. More significant are peripheral ulcerative keratitis and keratolysis (“corneal melt”) both of which have a high risk of perforation.2 Necrotizing keratitis or scleritis in the context of RA are associated with increased mortality.23
Posterior segment lesions in RA include posterior scleritis and rarely a retinal vasculitis. Retinal vasculitis is probably underdiagnosed. In one study of 60 patients with RA the rate of retinal vasculitis was 18% even in the absence of clinical features of retinal vasculitis.24 This study is supported by a number of case reports describing typical retinal vasculitis associated with fluorescein angiographic evidence of leakage and a single case of retinal exudation in the context of scleritis that the authors attribute to vasculitis.25,26
Treatment
Treatment of systemic disease
The goals of treatment in RA are to alleviate symptoms and to prevent tissue destruction (usually of joints) and loss of function. Permanent damage to the joints may occur early in the disease and thus best practice is now to treat early and aggressively. The 2008 recommendations of the American College of Rheumatology (ACR) advise a hierarchical approach based on disease activity (low, moderate or high) and duration of disease (<6 months, 6–24 months, and 24 months).27 In addition to appropriate anti-inflammatory pharmacological interventions (such as NSAIDs, intra-articular and oral corticosteroids), disease-modifying antirheumatic drugs (DMARDs) such as methotrexate and lefluonimide are recommended first-line and may be used in combination according to activity and duration of disease. The ACR recommendations for use of biologics (such as anti-TNF therapies) include high disease activity, poor prognostic features (such as the presence of rheumatoid nodules, secondary Sjögren syndrome, rheumatoid vasculitis, longer duration of disease, and failure of DMARDs).27 Anti-TNF therapy may also be used with methotrexate. Other biologic therapies include rituximab (anti-CD20) and abatacept (fusion protein of CTLA-4 and IgG).28
Treatment of ocular disease
Mild superficial ocular disease (such as mild keratoconjunctivitis sicca or episcleritis) may be adequately controlled with topical therapies such as artificial tear substitutes.29 Non-necrotizing anterior scleritis may be managed with oral NSAIDs, but necrotizing disease frequently requires systemic corticosteroid. Uncontrolled ocular inflammation warrants escalation of systemic treatment that should be coordinated with a rheumatologist. In addition, sight-threatening inflammation such as necrotizing scleritis or corneal melt requires urgent rescue therapy such as pulsed intravenous methylprednisolone.2 In our practice we administer up to three pulses of 500–1000 mg methylprednisolone on consecutive days that is usually followed by commencing or increasing a course of oral corticosteroid (in addition to DMARD/biologic therapy).
Seronegative spondyloarthropathies
General considerations
Spondyloarthropathy is a term used to describe a group of interrelated inflammatory arthropathies affecting the synovium and extra-articular sites (Box 80.2).30 The spondylarthropathies include the following conditions: ankylosing spondylitis, reactive arthritis, inflammatory bowel disease-related arthritis, juvenile spondyloarthropathies, and psoriatic arthritis. Clinical manifestations include inflammatory back pain, enthesitis (inflammation of the entheses, where tendons or ligaments insert into the bone), dactylitis (inflammation of an entire digit), uveitis, and usually an asymmetrical arthritis that affects lower limbs. There is a strong association with the class I MHC molecule HLA-B27. Based on a systematic review, which included nearly 30 000 patients, the mean prevalence of uveitis in spondylarthropathies has been estimated at 33% overall, with acute anterior uveitis being the most common type seen.31
Box 80.2
The European Spondyloarthropathy Study Group (ESSG) spondyloarthropathy classification criteria
(Reproduced with permission from Dougados M, van der Linden S, Juhlin R, et al. The European Spondylarthropathy Study Group preliminary criteria for the classification of spondylarthropathy. Arthritis Rheum 1991;34:1218–27.)
Epidemiology
Spondyloarthropathies occur particularly in individuals who are positive for HLA-B27 but additional environmental factors are also thought to play a role. It can be difficult to differentiate these disorders, because their clinical features may overlap and undifferentiated forms of spondyloarthropathy are well recognized.32 Spondyloarthropathies as a whole have a prevalence of 0.5–1.9% of the population.
Ankylosing spondylitis
Epidemiology
AS is the commonest of the spondyloarthropathies. Its prevalence varies between 0.1 and 0.4% of the population, depending on the frequency of HLA-B27 in that population. This results in significant geographic variation with extremely low rates of AS in South Africa, low rates in Japan, higher rates in Germany compared to other European countries and very high rates in the natives of Eurasia and the North American circumpolar/sub-Arctic areas.33 AS is commoner in males, with a male: female ratio of 2–3 : 1, although it has been suggested that it may be underdiagnosed in females due to them having milder disease.34
Articular and systemic disease
The hallmark of AS is inflammatory back pain presenting with buttock pain, early morning stiffness (minimum 30 minutes), relieved by exercise and NSAIDs, worse with rest, and night pain.35 The shoulders and hips are regarded as axial joints and are affected in up to 50% of patients.36 In AS an asymmetrical oligoarthritis is uncommon, but may be a predictor of more severe disease if it presents early in the disease course.36
Enthesitis is a characteristic feature of AS and may occur at any enthesis, but is most commonly seen in the foot at the insertion of the Achilles tendon and of the plantar fascia onto the calcaneus. The classic cardiac abnormalities in AS are aortitis, aortic regurgitation, and conduction abnormalities, which are seen in up to 9% of long-term follow-up patients.37
Ocular disease
The commonest ocular complication of AS is recurrent AAU. It is almost always unilateral but may affect both eyes sequentially (described as “flip-flop” pattern). Rarely the anterior uveitis may become persistent. The presentation is of typical AAU but inflammation is classically more severe (often with fibrin in the anterior chamber) and recurrences more frequent than in idiopathic AAU.38–40 Hyopyon may also be present in severe cases. AAU may lead to sight loss via recurrent or persistent CME, secondary glaucoma, and cataract.38–40 Treatment-related ocular complications of AS include cataract and elevated intraocular pressure (secondary to corticosteroid usage).
Treatment
Treatment of systemic disease
The goal of treatment in AS is to restore and maintain posture and movement to as near normal as possible, which is achieved through lifelong physical therapy, and medical and surgical treatment. NSAIDs (non-steroidal anti-inflammatory drugs) are the basis of treating AS. They reduce pain and stiffness within 48–72 hours in several studies; a good response may also be used to support the diagnosis.41,42 Unlike many inflammatory rheumatic diseases, systemic corticosteroids do not have a major part in the treatment of AS although peripheral arthritis often responds to steroid treatment.41
DMARDs (disease-modifying antirheumatic drugs), such as sulfasalazine and methotrexate, are effective for the peripheral manifestations of AS, but there is limited efficacy for treating axial manifestations.43,44 Anti-TNF agents, such as etanercept, infliximab, and adalimumab are recommended as treatment options for patients with AS if disease is not sufficiently controlled after treatment with two or more DMARDs.
Treatment of ocular disease
The mainstay of treatment for AS-associated AAU is intensive topical corticosteroids and a mydriatic (as for idiopathic AAU), but it should be noted that subconjunctival treatment and oral corticosteroids are more commonly required to adequately control inflammation in HLA-B27-associated versus idiopathic AAU. Anti-TNF agents, such as infliximab, etanercept, and adalimumab, as part of studies for the treatment of the underlying AS can also reduce the frequency of AAU recurrences.45,46
Reactive arthritis (previously known as Reiter syndrome)
Epidemiology
There are a few population-based studies which have estimated the incidence to be between 10 and 30 per 100 000 per annum.47 HLA-B27 is positive in 60–80% of patients with reactive arthritis. The most commonly associated infections are urogenital (Chlamydia trachomatis) or gastrointestinal (Yersinia, Salmonella, Shigella and Campylobacter).48–50
Articular and systemic disease
Characteristically patients will have an asymmetric oligoarthritis of the large lower limb joints, but the upper limbs and small joints (usually PIP joints rather than MCP joints) are affected. Other features include inflammatory lower back pain similar to AS, dactylitis, enthesitis, erythema nodosum, and keratoderma blenorrhagica (pustular skin lesions on the soles of feet), indistinguishable from pustular psoriasis.48
Ocular disease
The commonest ocular complications of ReA are anterior segment disease. Conjunctivitis forms part of the classical triad of ReA, but is usually only seen at first presentation and early in the disease . Of more consequence is recurrent AAU that may occur in up to 50% of patients, although in only up to 20% patients at the initial attack.40,48 As described for other forms of AAU, these attacks may be associated with spillover vitreous cells and CME, and very rarely optic disc edema.
Rarely ReA may be associated with panuveitis or multifocal choroiditis;51,52 the rarity of such cases is underlined by many series of ReA patients in which no cases of panuveitis or posterior uveitis were identified.48,53
Treatment
Treatment of systemic disease
The underlying infection should be treated as appropriate.54 Acute arthritis can be treated with NSAIDs and intra-articular corticosteroids. DMARDs such as sulphasalazine or methotrexate should be considered in patients with a prolonged disease course.54
Inflammatory bowel disease
Epidemiology
The incidence of peripheral arthritis is reported to be between 5% and 10% in ulcerative colitis and 10% and 20% in Crohn’s disease, respectively.55 Men and women are affected equally. Spondylitis occurs in 1–26% of patients with IBD and males are more often affected than females. In addition, the prevalence of AS in IBD (1–6%) is higher than in the general population.55
Articular and systemic disease
IBD-related arthritis is often a clinical diagnosis as radiology is often normal with no joint erosion or deformity. Two distinct types of arthritis have been described: type 1 (pauciarticular) and type 2 (polyarticular).55,56
Ocular disease
Ophthalmic complications are estimated to occur in 3.5–12% of patients with IBD, occurring more commonly earlier in the disease.57 The commonest ocular complications of IBD are uveitis, episcleritis, and scleritis.
Uveitis is usually of recurrent AAU type, occurring in around 5% of IBD patients, but in up to 50% of IBD patients who are also positive for HLA-B27.58 Less commonly, a chronic bilateral anterior uveitis may be seen, which has a female gender preponderance.59 Scleritis is a well-recognized feature of IBD and has been reported to parallel the activity of the bowel disease. Scleritis is usually anterior but may be posterior; it may be non-necrotizing or necrotizing, leading to a risk of scleromalacia perforans.60 Retinal artery occlusions and ischemic optic neuropathy are reported and may reflect the prothrombotic tendency seen in some patients with IBD. Other reported associations with IBD include keratitis, retinal vasculitis, posterior uveitis, cystoid macular edema, optic neuritis, neuroretinitis, Brown’s syndrome and orbital myositis.60 A recent community survey also suggested that there is a high prevalence of “dry eye” (up to 42%), which was associated with 5-aminosalicylate use, although it was not clear if this was causative or a surrogate marker of disease activity.61 Interestingly, a family history of IBD has been proposed as an independent risk factor for the development of idiopathic ocular inflammation, including uveitis.62
Treatment
Treatment of systemic disease
Treatment depends on the severity of symptoms. Patients with mild oligoarthritis usually respond to relative rest, physiotherapy, and intra-articular corticosteroid injections.55,56 Most of the patients respond to NSAIDs because they control the symptoms and joint and enthesis inflammation, but they do not stop joint destruction, and they may have significant side-effects including exacerbation of IBD and produce small intestine and colon ulcers. Hence, they are recommended for patients with mild exacerbations, to control symptoms in arthritis flares, but their use must be limited to the minimal effective dose and time.55,56
Type 1 arthritis is related to disease activity and therefore therapy of the underlying IBD is the treatment of choice. Treatment of type 2 IBD arthritis and axial arthropathies generally requires long-term treatment with a DMARD, such as sulfasalazine or methotrexate. In addition treatment with systemic on intra-articular corticosteroids can be used.55 A number of IBD patients will also be using anti-TNF agents to control their bowel symptoms.
Treatment of ocular disease
IBD-associated AAU is treated as for idiopathic AAU with intensive topical corticosteroids and a mydriatic; it should be noted that the more chronic form of anterior uveitis may require more prolonged treatment.63 The treatment of scleritis will depend on the pattern and severity of disease seen, but will often require immunosuppression.63
Psoriatic arthritis
General considerations
Psoriatic arthritis (PsA) is the combination of an inflammatory arthritis (peripheral arthritis and/or sacroiliitis or spondylitis), psoriasis, and the absence of serological tests for rheumatoid factor.30 In 2006, however, the CASPAR (ClASsification Criteria for Psoriatic ARthritis) was developed (Box 80.3). This has been demonstrated to have high sensitivity and specificity for the diagnosis of PsA (see below).30,64
Box 80.3
The CASPAR criteria for psoriatic arthritis
At least 3 points scored from the following
(Adapted with permission from Taylor W, Gladman D, Helliwell P, et al. Classification criteria for psoriatic arthritis: development of new criteria from a large international study. Arthritis Rheum 2006;54:2665–73.)
Articular and systemic disease
PsA can cause a variety of articular symptoms, varying from an isolated monoarthritis to an extensive destructive arthritis. Articular involvement can be divided into five subtypes: DIP (distal interphalangeal) joint involvement, mono/oligoarticular, symmetrical polyarthritis, arthritis mutilans, and spondylarthropathy. It is important that patients with longstanding PsA have cervical spine X-rays before a general anesthetic, as there can be a clinically silent erosive/inflammatory arthritis causing atlantoaxial or subaxial instability, as in RA.67 Dactylitis or “sausage digit” occurs in 30–40% of patients with PsA. In addition 20–40% of patients have symptomatic enthesitis generally affecting the foot at the insertion of the Achilles tendon and of the plantar fascia onto the calcaneus.64,68 There appears no correlation between the severity of skin psoriasis and joint involvement, but skin symptoms do tend to precede joint symptoms. Psoriasis can be assessed using a variety of tools including: PASI (Psoriasis Area and Severity Index), health assessment questionnaires, and Psoriatic Arthritis Response Criteria (PsARC).
Ocular disease
Ophthalmic complications are estimated to occur in 10% of patients with psoriasis,69 and 31% of patients with PsA.70 The most common presentations are conjunctivitis (up to 20%) or uveitis (7%).70 Paiva and coworkers compared the nature of uveitis in PsA to that seen with a previous cohort of spondylarthropathy patients. Interestingly this suggested that PsA-associated uveitis was more likely to be insidious in onset (19% versus 3%), bilateral (38% versus 7%), chronic in duration (31% versus 6%) or posterior (44% versus 17%).71 Episcleritis, scleritis, keratoconjunctivitis sicca, and keratitis are also reported.69,70
Treatment
Treatment of systemic disease
NSAIDs are normally used for musculoskeletal symptoms, based on the evidence from other rheumatic diseases.72 Intra-articular corticosteroid can be used, but oral corticosteroids should be used cautiously as they can be associated with a “post-steroid psoriasis flare.” Methotrexate can be used for both skin psoriasis and PsA, but care must be taken as the risk of hepatotoxicity seems to be higher in patients with psoriasis, possibly due to a higher tendency of non-alcoholic steatohepatitis (NASH). Sulfasalazine can be used for articular symptoms; there is no benefit for the skin psoriasis. Leflunomide has demonstrated to be effective at treating PsA.72 Cyclosporine can achieve rapid improvement of the skin lesions of psoriasis, but there is little evidence regarding its effectiveness in PsA. There are concerns regarding possibly causing hypertension and renal insufficiency, so the dose needs to be kept as low as possible and careful monitoring is indicated as these reversible events are reversible if picked up soon after onset.72
Anti-TNF agents (etanercept/infliximab/adalimumab) are generally reserved for severe disease. They have been shown to be effective at treating peripheral arthritis, psoriasis, enthesitis, and dactylitis.73 Further work is currently being undertaken to look at other possible biologic targets including: alefacept, which is a fully human fusion protein that blocks interaction between LFA-3 on the antigen-presenting cell; rituximab, an anti-CD20 agent; and ustekinumab, an IL-12/IL-23 inhibitor.73
Treatment of ocular disease
PsA-associated AAU is treated as for idiopathic AAU with intensive topical corticosteroids and a mydriatic, but more persistent anterior uveitis may require prolonged treatment. Cataract, which may be associated with chronic intraocular inflammation, corticosteroid usage, and possibly P-UVA treatment,74 requires surgical treatment with appropriate immunosuppressive perioperative care.
Juvenile idiopathic arthritis
Epidemiology
The reported incidence of JIA varies between 0.8 to 23 per 100 000 per annum, with a prevalence rate between 7 and 400 per 100 000 children. There are reported differences between different ethnic groups: JIA is more frequent in children of European descent than in children of African, Asian or East Indian origin.75
Articular and systemic disease
JIA is classified using the ILAR (International League of Associations of Rheumatologists) classification (Box 80.4). The main clinical feature of JIA is defined as: “swelling within a joint or limitation in range of movement with joint pain or tenderness, which persists for a minimum of 6 weeks, observed by a physician and which is not due to primary mechanical disorders or to other identifiable causes.”76
Box 80.4
The International League of Associations of Rheumatologists (ILAR) classification of juvenile idiopathic arthritis
Psoriatic arthritis
Definition: Arthritis and psoriasis, or arthritis and at least two of the following:
Enthesitis-related arthritis
1. The presence of or a history of sacroiliac joint tenderness and/or inflammatory lumbosacral pain
2. The presence of HLA-B27 antigen
3. Onset of arthritis in a male over 6 years of age
4. Acute (symptomatic) anterior uveitis
5. History of ankylosing spondylitis, enthesitis-related arthritis, sacroiliitis with inflammatory bowel disease, Reiter syndrome, or acute anterior uveitis in a first-degree relative
Exclusions
a. Psoriasis or a history of psoriasis in the patient or first-degree relative
b. Arthritis in an HLA-B27-positive male beginning after the 6th birthday
c. Ankylosing spondylitis, enthesitis-related arthritis, sacroiliitis with inflammatory bowel disease, Reiter syndrome, or acute anterior uveitis, or a history of one of these disorders in a first-degree relative
d. The presence of IgM rheumatoid factor on at least two occasions at least 3 months apart
(Reproduced with permission from Petty RE, Southwood TR, Manners P, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol 2004;31:390–2.)
Ocular disease
The major ocular manifestation of JIA is uveitis. Uveitis is present in around 10% of JIA patients at presentation but may occur in up to one-third of patients at some point during their disease although the exact estimate depends on the type of population sampled.77–81 Inflammation is typically a chronic anterior uveitis with a white eye, which is usually bilateral (70%) but may initially present with unilateral disease. Recurrent acute anterior uveitis is less commonly seen and when it does occur is usually in the context of HLA-B27.
The risk of developing uveitis in association with JIA has been stratified according to age of onset, type of arthritis, and the presence of ANA, with uveitis occurring in up to half of the highest-risk group (oligoarticular – persistent and extended, ANA-positive disease). Interestingly, a recent study has suggested that age and ANA status may be less useful in predicting risk in boys (versus girls).82 It should be noted that the gold standard for determining ANA is via immunofluorescence on HEp-2 cells; ELISA-determined ANA was not found to be predictive.83 Antihistone antibodies are also associated with risk of uveitis but are less widely used in clinical practice.83 Since the chronic anterior uveitis has minimal if any of the symptoms commonly associated with inflammation, screening is recommended. The recommendations of the American Academy of Pediatrics are summarized in Table 80.1.84
Table 80.1 American Academy of Pediatrics guidelines on frequency of ophthalmologic examination in patients with juvenile idiopathic arthritis
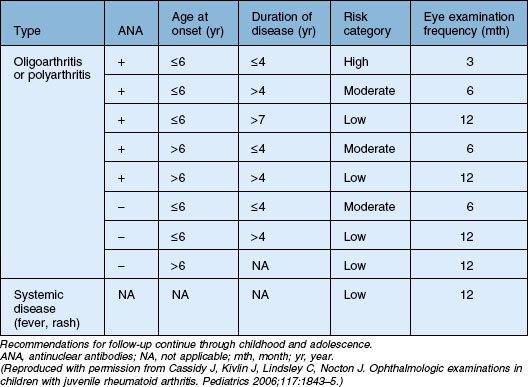
JIA-associated uveitis is more commonly seen in girls,81 but male gender is a risk factor for worse disease, with significantly higher rates of CME by 5 years of follow-up (50% versus 4%) and need for cataract surgery (59% versus 32%). Other reported predictors of worse outcome are uveitis present at time of diagnosis85,86 and elevated laser flare values.87
The key sight-threatening complications of JIA-associated uveitis are band keratopathy (60%), cataract (40%), glaucoma (10–25%), and CME (10%). Posterior synechiae are present in most cases.77,88,89 Less commonly, vitritis and a peripheral retinal vasculitis are reported.77,80,88,89
Treatment
Treatment of systemic disease
The management of JIA is based on a combination of medical treatment, physical and occupational therapy, and surgical management. NSAIDs can be used for all types of mild JIA, to treat pain and stiffness. Oral corticosteroids must be used minimally in children because of the effects on bone and growth, the main indications are severe fever, serositis and macrophage activation syndrome. Intra-articular corticosteroids can be used, and encouraging results have been reported in children with monoarthritis.90 Methotrexate is used for patients with polyarthritis, other DMARDs that have been shown to be effective include sulfasalazine and leflunomide. Etanercept can be used in patients who fail to respond to methotrexate therapy and has been demonstrated to be effective both in the short and long term management of JIA.91–93 Infliximab, was not shown to be superior to placebo in polyarticular JIA.94 Adalimumab is licensed for use in JIA, and was found to be effective in children with polyarticular JIA.95 Abatacept is an alternative biological agent which is a selective T-cell co-stimulator inhibitor. In randomized controlled trials (RCTs) abatacept was superior to placebo in children with polyarticular arthritis, including those who were anti-TNF treatment failures.96,97 There is some concern regarding the potential increased risk of cancers in children using anti-TNF agents.98 Further studies are being undertaken to investigate the potential future role of other biologics, including rituximab, IL(interleukin)-1 and IL-6 antagonists.
Treatment of ocular disease
Systemic immunosuppression is usually required to control both the systemic disease and its ocular manifestations, although some children may only require topical corticosteroid and a mydriatic. What constitutes a “safe” level of topical corticosteroid usage in children is controversial. In a recent retrospective study by Thorne and coworkers topical corticosteroid use was associated with development of cataract and this was independent of active uveitis or presence of posterior synechiae.99 Importantly, there appeared to be no significant increase of cataract when chronic administration was no more than twice daily.99 The need for frequent topical corticosteroid to control the uveitis usually requires the introduction of methotrexate often given by the subcutaneous route weekly rather than orally. If methotrexate cannot adequately control the inflammation then it requires the addition of an anti-TNF agent; adalimumab appears to be the drug of choice, with effective control of the uveitis in 16/18 children in one study.100 There are some reports of uveitis occurring in association with etanercept.101
Cataract surgery is challenging and requires very careful preparation, perioperative care and intensive postoperative management. It is vital that the patient’s carers are fully aware of the importance of adherence to prescribed therapy and follow-up visits. Traditionally cataract removal for these patients was by pars plana vitrectomy/lensectomy followed by aphakia, but it is now more common to use intraocular lenses at the time of surgery.102–104 Postoperative posterior synechiae and intraocular lens deposits are common, but overall visual improvement is encouraging, with a recent series of 17 eyes reporting an improvement in visual acuity of 2 lines or more in all patients with no increase of CME, glaucoma or hypotony,103 but it should be noted that other studies do report significant rates of secondary glaucoma and CME.104
Systemic lupus erythematosus
General considerations
Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease predominantly affecting women of childbearing age. The classification criteria for SLE are summarized in Box 80.5.105
Box 80.5
The 1997 updated American College of Rheumatology criteria for systemic lupus erythematosus
Fixed erythema, flat or raised, over the malar eminences, tending to spare the nasolabial folds
Skin rash as a result of unusual reaction to sunlight, by patient history or physician observation
Oral or nasopharyngeal ulceration, usually painless, observed by physician
Involving 2 or more peripheral joints, characterized by tenderness, swelling, or effusion
At least one of the following:
At least one of the following:
11. Positive antinuclear antibody
(Modified from Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997;40:1725.)
Epidemiology
SLE has a prevalence of around 28 cases per 100 000,106 predominantly affecting women of child-bearing age (15–45 years); the female: male ratio peak is 12 : 1. SLE is more common in non-Caucasians, in whom it is both more severe and earlier in onset.107
Articular and systemic disease
SLE is a systemic disease that can cause constitutional or organ-specific symptoms. The skin, mucous membranes, joints, kidney, brain, serous membranes, lung, heart, and occasionally the gastrointestinal tract may all be involved.105,106 Arthritis in SLE can divided into a deforming and nondeforming arthropathy. Constitutional symptoms consist of fever, malaise, fatigue, weight loss, lymphadenopathy, and anorexia.106 There are multiple cutaneous manifestations of lupus: commonly these include photosensitivity (>50% of patients), butterfly/malar rash, painful/painless oral ulcers, diffuse alopecia, and livedo reticularis.106
Renal disease is one of the most serious SLE manifestations and a prognostic indicator of disease severity. Renal biopsy is used to determine the class of nephropathy as classified by the International Society of Nephrology (ISN)/Renal Pathology Society (RPS) guidelines.108
Neuropsychiatric SLE (NPSLE) is a major diagnostic and treatment problem. The ACR has provided classification criteria, describing central and peripheral types of neurological involvement that may be found in lupus patients.106
Pulmonary features of SLE include pleurisy, pneumonitis, pulmonary hemorrhage, pulmonary embolism, pulmonary hypertension, and diaphragmatic weakness causing shrinking lungs. Pericarditis is the most common cardiological manifestation; others include myocarditis, endocarditis, accelerated atherosclerosis, and, rarely, pericardial tamponade.106
Abdominal pain, nausea, vomiting, and diarrhea occur in up to 50% of SLE patients. Gastrointestinal involvement includes mesenteric vasculitis (high risk of death), aseptic peritonitis (with or without ascites), subacute bowel obstruction, hepatitis, sclerosing cholangitis, protein-losing enteropathy, pancreatitis, and ascites.109
Cytopenias, including anemia, leucopenia or thrombocytopenia, are commonly associated with SLE; they may be immune-mediated or due to other factors, e.g. menstrual losses. Antiphospholipid antibodies and lupus anticoagulant are found in about 30–40% of patients, associated with venous and arterial thrombosis, recurrent fetal loss, pre-eclampsia, headache, and epilepsy.106
A firm diagnosis of lupus is made based on appropriate clinical findings and the measurement of at least one antibody. There are a number of antibodies associated with SLE, the most common being ANA (antinuclear antibody). Other associated autoantibodies include anti-dsDNA (double stranded DNA) in approximately 60% of patients, the highly specific anti-Sm antibody (Smith proteins) in 10–30% of patients, and anti-RNP (ribonucleoprotein) also in 10–30% of patients.105,106
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


