Retinal Vasculitis
Margaret Wong
Fernandino A. Fontanilla
Debra A. Goldstein
Ozlem Sahin
Howard H. Tessler
Retinal vasculitis may occur alone or as part of a systemic disease and may be the first manifestation of life-threatening illness. The ophthalmologist must ensure that investigations are obtained to determine whether the retinal vasculitis exists alone or as part of a systemic illness.
IMMUNE MECHANISMS OF VASCULITIS
IMMUNE COMPLEXES
Immune complexes are formed by the association of an antibody with an antigen. Low levels of circulating immune complexes are found in most people and may promote the efficient removal of tissue debris or excess antigen. Immune complexes can activate the complement cascade, attracting polymorphonuclear leukocytes that release proteolytic enzymes, and cause tissue or vascular injury.
Circulating immune complexes and complement abnormalities have been reported in connective tissue diseases such as systemic lupus erythematosus (SLE) and polyarteritis nodosa, in Behçet disease, in HLA-B27+ uveitis, and in idiopathic retinal vasculitis.1,2,3 The Arthus reaction is a model for immune complex vasculitis and produces histopathologic changes resembling those seen in Behçet disease.4 Because of these findings, retinal vasculitis was postulated to be caused by immune complex deposition.5 However, the role of immune complex-mediated tissue damage in the eye remains unclear. Studies on idiopathic retinal vasculitis suggest that immune complexes may actually have a protective function, neutralizing antiretinal autoantibodies.6,7
AUTOANTIBODIES
Antibodies may bind directly to surface antigens of cells and tissues, leading to activation of the complement system and effector cells, resulting in cell lysis or cytotoxic damage.8 An example of antibody-mediated ocular disease is cancer-related retinopathy (CAR), in which antibodies that are produced against a tumor cross-react with retinal tissue, causing retinal damage.9
Antibodies to human vascular endothelial cells have been detected in the sera of patients with various vasculitic disorders, including Wegener granulomatosis and polyarteritis nodosa.10,11,12,13 Autoantibodies to endothelial cells were found in 47% of patients with retinal vasculitis associated with systemic disease and in 35% of patients with idiopathic retinal vasculitis.14 In the same study, only 1% of normal controls had anti–endothelial cell antibodies. Possible mechanisms by which they induce vascular damage include complement fixation, neutrophil recruitment, and antibody-dependent cellular cytotoxicity.15 In Behçet disease, anti–endothelial cell antibodies have been associated with systemic thrombotic complications.14
Anti–neutrophilic cytoplasmic autoantibodies have been detected in patients with Wegener granulomatosis. It is postulated that these antibodies interact with stimulated neutrophils, resulting in their activation.16 The activated neutrophils then adhere to vascular endothelia and undergo degranulation, generating oxygen radicals that result in endothelial cell injury and inflammation.17 In vitro observations also indicate that anti–neutrophilic cytoplasmic autoantibodies may interact directly with endothelial cells.16
OTHER MECHANISMS
Delayed hypersensitivity plays a role in Wegener granulomatosis, sarcoidosis, and sympathetic ophthalmia.8 In sarcoidosis, the macrophage is thought to initiate the inflammatory response.18 Abnormal cell-mediated immune responses to photoreceptor antigens have been found in idiopathic retinal vasculitis,19 and patients with retinal vasculitis have also been shown to have higher levels of natural killer cell activity.20
GENETIC PREDISPOSITION
The major histocompatibility complex plays an important role in the initiation of the immune response. HLA-DR3 is common in SLE and Sjögren syndrome, and HLA-DR4 is common in idiopathic retinal vasculitis, rheumatoid arthritis, and Takayasu disease.21 Certain HLA types have also been linked to Behçet disease (B51), multiple sclerosis (DR2), and birdshot retinochoroidopathy (A29).22,23,24
CLINICAL FEATURES AND PATHOPHYSIOLOGY
Inflammation of the retinal vessels is seen clinically as white cell cuffing, sheathing, or perivascular exudation. Patients with retinal vasculitis typically describe a painless decrease in vision, which may be associated with floaters or vitreous hemorrhage. Large areas of visual field loss, possibly owing to ischemia, may also be present. Peripheral retinal involvement may result in minimal symptoms, but posterior pole involvement may be more symptomatic. In many patients, anterior uveitis, vitritis, and chorioretinitis accompany the vasculitis.
PERIPHLEBITIS AND PHLEBITIS
Venous inflammation is much more common than arteritis. Early venous changes include patchy dilatation, venous irregularity, and perivenous cuffing. These cuffs are made up of white blood cells and vary greatly in density, from minimal obscuration of a vein to complete concealment of the blood column without occlusion. Vitreous cells may be present over the vessels. Vascular sheathing appears as white lines along the vein walls and may result in venous flow impedance and vein occlusion (Figs. 47-1 and 47-2).25 It may be clinically difficult to distinguish perivenous inflammation from true phlebitis. Any vein from the optic nerve head to the peripheral retina is susceptible.
 Figure 47.1. Linear venous sheathing in a patient with Eales disease. |
 Figure 47.2. Patient with Behçet disease. Notice the linear sheathing of the superotemporal vein with adjacent retinal hemorrhage. There is intermittent sheathing along the superonasal vessels. |
If inflammation is prolonged, secondary changes such as thickening of the vein wall and endothelial cell proliferation may occur. These can lead to narrowing and obstruction of the lumen, thrombosis, and necrosis. Other late changes secondary to vascular occlusion include telangiectasia, microaneurysms, and neovascularization.21 Histopathologically, polymorphonuclear leukocytes are the predominant cell type early in the disease, but lymphocytes accompanied by an occasional plasma cell, giant cell, or epithelioid cell become the predominant cell types later in the disease.25
Fluorescein angiography often reveals focal areas of staining or dye leakage before ophthalmoscopically visible signs are present (Fig. 47-3). In advanced cases of periphlebitis, angiography may show diffuse leakage of dye from retinal veins and capillaries.26
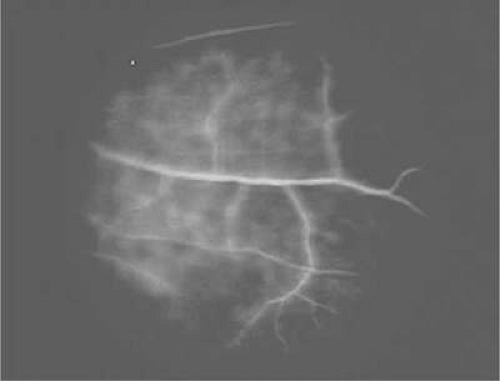 Figure 47.3. Fluorescein angiogram of a patient with systemic lupus erythematosus showing perivenous staining indicative of phlebitis. |
ARTERITIS AND PERIARTERITIS
The arterial changes in retinal vasculitis are varied. Immune complex deposition in precapillary arterioles may result in cotton-wool spots, as seen in SLE and certain infectious conditions.27,28 Aneurysmal dilatation of arteries has been observed in various infections, connective tissue diseases, and hypersensitivity vasculitides (Fig. 47-4).29
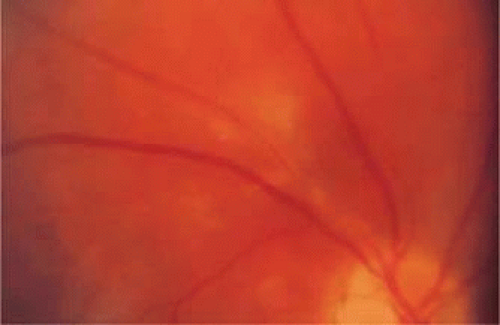 Figure 47.4. Sheathing and inflammation around the superotemporal arteriole in a patient with idiopathic retinal vasculitis. |
Arterioles may show segmental, periarteriolar, hard, yellowish nodular plaques (kyrieleis arteriolitis) that do not extend beyond the thickness of the vessel wall (Fig. 47-5). The plaques are not visible angiographically and do not alter dye transit or cause permeability changes. Primarily associated with toxoplasmosis, they have also been seen in syphilis, tuberculosis, cytomegalovirus, and herpes zoster retinopathy.30,31,32
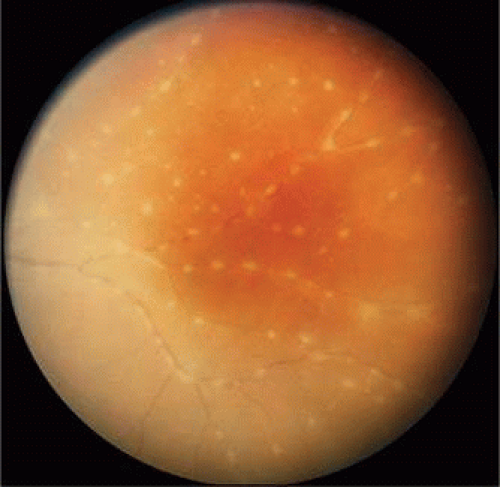 Figure 47.5. Kyrieleis vasculitis in a patient with active toxoplasmosis. The vasculitis resolved with no sequelae after treatment of the toxoplasma lesion (not shown). |
Severe inflammation can result in dense irregular sheathing of large arterial segments. This sheathing can conceal the blood column and extend beyond the width of the vessel wall. As the inflammation subsides, the arteries become white ghost vessels. Fluorescein angiography shows arteriolar occlusion with surrounding capillary nonperfusion. After arteriolar occlusions, neovascularization may occur in the areas of nonperfused retina (Fig. 47-6). This picture has been seen in connective tissue diseases, primary vasculitic diseases, necrotizing viral retinitis, and Eales disease.33 Pigment proliferation in a bone spicule pattern may also occur around and beyond the occluded arterioles.
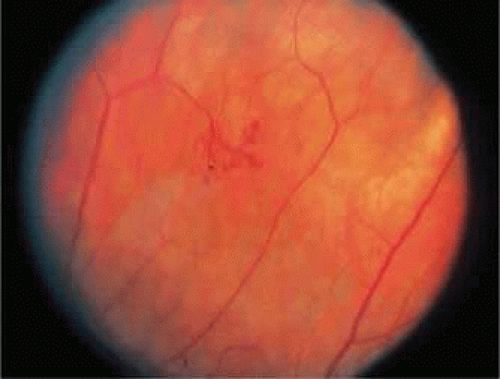 Figure 47.6. Sea fan–like neovascular frond formed after vascular occlusion in a patient with Behçet disease. The patient did not have diabetes or sickle cell disease. |
LOCAL RETINAL VASCULITIS
EALES DISEASE
Eales disease was initially described as recurrent retinal and vitreous hemorrhages, epistaxis, constipation, and headaches in young men.34 It has since been redefined as an idiopathic obliterative peripheral retinal vasculopathy with variable degrees of nonperfusion, vascular sheathing, retinal vascular abnormalities, peripheral retinal neovascularization, and hemorrhage (Fig. 47-7).35 It usually affects men in the third decade of life and is bilateral in up to 90% of cases.36 Many patients are symptomatic in only one eye, but fundus examination of the fellow eye may reveal early changes such as periphlebitis, vascular sheathing, and peripheral retinal nonperfusion.37 The presenting symptom in 90% of patients is a painless blurring of vision, often caused by vitreous hemorrhage.36
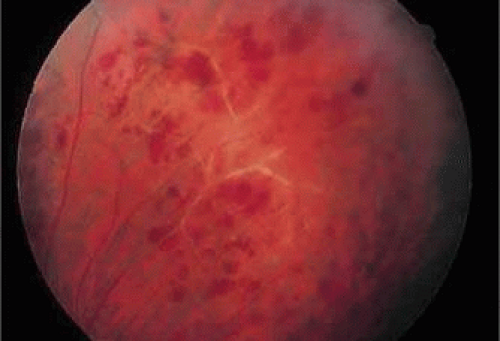 Figure 47.7. Patient with Eales disease. Note the vascular sheathing, retinal hemorrhages, and peripheral venous obliteration. The patient later developed sarcoidosis. |
The clinical manifestations of Eales disease result from three basic pathologic changes: inflammation, ischemia, and neovascularization.38 Vitreous cells are present and anterior segment inflammation is variable. In early stages, venous dilatation and tortuosity are seen in the periphery.37 Accumulation of exudate around peripheral retinal venules appears as thin white lines parallel to the blood column and can obscure the vessel. Areas of vascular sheathing frequently leak fluorescein dye; however, the sheathing does not always correspond to the staining, nor is the amount of leakage proportional to the activity of the inflammation.37
Progression of Eales disease results in widespread venous occlusion, perivenular or arteriolar exudation, extensive sheathing, and retinal hemorrhage. Involved vessels become obliterated, and avascular areas develop in the periphery. The junction between nonperfused and perfused retina is usually sharply demarcated.37 Vascular abnormalities seen at this junction include microaneurysms, venovenous shunts, venous beading, hard exudates, and cotton-wool spots.39,40 Obliteration of peripheral venules and arterioles and branch retinal vein occlusions may be confirmed angiographically.41
Capillary nonperfusion leads to peripheral and disc neovascularization; this is observed in up to 80% of patients with Eales disease37 and contributes to the development of fibrous proliferation and the risk of retinal detachment. Peripheral neovascularization is more common and is frequently located at the junction between perfused and nonperfused retina. Bleeding from damaged or abnormal vessels may result in vitreous hemorrhage. Extension of nonperfusion into the posterior pole may result in decreased visual acuity owing to macular ischemia or edema, epiretinal membrane, or macular hole formation. Patients may also develop anterior uveitis, cataract, rubeosis iridis, secondary neovascular glaucoma, and optic atrophy in the late stages of the disease.37
There is histopathologic evidence of lymphocytic and granulomatous infiltration of vessel walls and lumina, and perivascular spaces.42 The cause of Eales disease, however, is unknown. It may represent a hypersensitivity reaction to tuberculoprotein. This hypothesis stems from isolated reports of vasculitis after skin testing or BCG vaccine and the prevalence of positive purified protein derivative testing in patients with the disease.43 Tubercles in the venous walls were also reported in the older literature.44 One study found elevated IgA and IgG levels in patients with Eales disease,45 whereas other studies showed no immunoglobulin abnormality.46,47 One study described vascular endothelial growth factor (VEGF) in the vitreous of Eales patients, possibly explaining the neovascularization.48 A case report of one patient whose retinal neovascularization resolved following intravitreal injection of the anti-VEGF agent bevacizumab provides support for the VEGF finding.49 Abnormal levels of circulating immune complexes have also been reported.50 Eales disease probably has more than one cause.
IDIOPATHIC RETINAL VASCULITIS
Retinal vasculitis occurring in isolation, with no systemic association, has been referred to as idiopathic retinal vasculitis.51 Typical findings include inflammatory cells in the vitreous and sheathing of retinal veins and postcapillary venules. Fluorescein angiography may show diffuse capillary leakage.52 Idiopathic retinal vasculitis can be classified into ischemic and nonischemic forms. Patients with ischemic retinal vasculitis have a worse visual outcome despite aggressive systemic treatment than those with nonischemic retinal vasculitis.51 A few patients with idiopathic retinal vasculitis have disseminated central nervous system lesions characteristic of multiple sclerosis.52 Lyme disease and catscratch disease (Bartonella henselae) have been anecdotally associated with retinal vasculitis.53,54,55
OPTIC DISC VASCULITIS
Optic disc vasculitis (papillophlebitis) is a unilateral, idiopathic, usually benign condition that primarily affects healthy adults younger than 40 years of age.56 The only symptom is blurred vision. Retinal findings include edema of the optic disc and adjacent retina, markedly dilated and tortuous retinal veins with minimal arterial involvement, and a variable amount of retinal hemorrhage (Fig. 47-8). Other findings include venous sheathing, retinociliary shunt vessels, and depigmentation of the macular area.57 Vitreous cells are absent or scant. Fluorescein angiography findings may include delayed venous filling, marked retinal venous dilatation, and massive dye leakage from the optic nerve head and large retinal veins.56
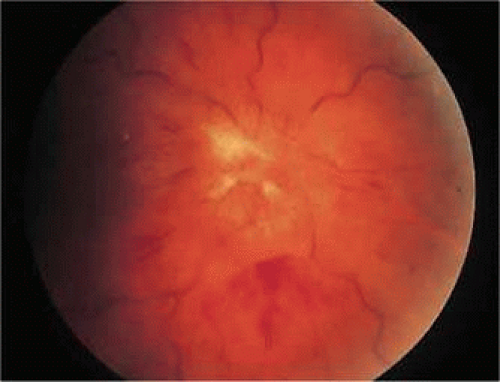 Figure 47.8. Young woman with idiopathic papillophlebitis. The optic nerve is markedly swollen, and the retinal veins are dilated and tortuous. Retinal hemorrhages are also seen. The patient did not improve with corticosteroids. She was found to have lupus anticoagulant, and vision improved from 20/800 to 20/50 with anticoagulation. |
Hayreh56 divided the disease into two types: type 1, with optic disc edema and good visual outcome as the dominant features, and type 2, with a clinical picture similar to central retinal vein occlusion. In the second type, the visual outcome depends on the site and extent of involvement. Hayreh56 suggested that type 1 is caused by a mild nonspecific vasculitis of the ciliary vessels, whereas type 2 is probably secondary to phlebitis of the central retinal vein within the optic nerve head or retrolaminar region.
Papillophlebitis is self-limited, usually lasting 6 to 18 months. Unlike peripheral retinal vasculitis (Eales disease), optic disc vasculitis is rarely recurrent or associated with vitreous hemorrhage. No therapy is necessary, although corticosteroids have been reported to shorten the course. There is controversy over the existence of papillophlebitis, and some believe it represents a mild central retinal vein occlusion in a young person.58
INTERMEDIATE UVEITIS
Intermediate uveitis is a term used to describe inflammation centered in the vitreous. Mild periphlebitis and optic disc and macular edema may also occur. Intermediate uveitis includes but is not synonymous with pars planitis. It may be idiopathic or associated with systemic disease such as sarcoidosis, multiple sclerosis, Whipple disease, and lymphoma.59 Pars planitis is an idiopathic intermediate uveitis that occurs in children and young adults. It is characterized by the presence of vitreous cells, snowballs, and exudates (snowbanking) over the inferior pars plana. Retinal vascular involvement has been reported in 13% to 77% of cases of pars planitis.60,61 Peripheral retinal veins may appear dilated, segmented, and tortuous, some with vascular sheathing. Retinal vascular leakage is common and was found in 79% of fluorescein angiograms in one study.60 Patchy perivenous staining in the posterior pole may accompany the closely spaced spots of fluorescein stain along peripheral venules. Arterial involvement has been reported infrequently.62 The most common cause of visual loss is cystoid macular edema. Vein occlusions, peripheral and disc neovascularization, vitreous hemorrhage, and retinal detachment are rare, but may occur.63,64 Histopathologic examination reveals thickening of the vessel wall and perivascular round cell cuffing.65 These changes can be observed as far posteriorly as the optic nerve.
ACUTE RETINAL NECROSIS
Acute retinal necrosis (ARN) is predominantly a fulminant retinitis with an associated obliterative vasculitis of the retina.66 Acute retinal necrosis is characterized by panuveitis, vitritis, vaso-occlusive retinal arteritis, and necrotizing retinitis.67 The most common cause of ARN is herpes zoster.68 Herpes simplex is also a common cause, particularly in younger patients, although other organisms such as cytomegalovirus (CMV) have also been reported.68 Vitreous haze often makes fundus examination difficult, but frequently arteriolar occlusions can be seen associated with areas of retinitis. There is an abrupt border between normal and abnormal retina, suggestive of an ischemic process. Retinal necrosis is usually located in the periphery of the retina, but a case series reported a variant in which the retinal necrosis was seen in the posterior pole.69 Progression results in retinal artery narrowing and sheathing. Perivenous sheathing and intraretinal hemorrhage may also be present. Angiographically, there is arteriolar obliteration and absence of capillary perfusion in affected areas, as well as dye leakage and vessel wall staining in the late phase.
FROSTED BRANCH ANGIITIS
Frosted branch angiitis, an inflammatory condition of unknown origin, is characterized by widespread periphlebitis with intraretinal hemorrhage in otherwise healthy patients. It is an acute condition and may be unilateral or bilateral. Fundus examinations reveal severe white sheathing of retinal vessels resembling frosted tree branches. Although both arteries and veins may be involved, the venules tend to be more commonly affected. The inflammatory exudates around retinal vessels may be related to deposition of antigen-antibody complexes.75,76 Anterior chamber and vitreous inflammation may be seen. On fluorescein angiography, there is late staining and dye leakage with no evidence of stasis or occlusions.21 Frosted branch angiitis has also been reported in patents with cytomegalovirus retinitis (Fig. 47-9)77 and toxoplasmosis.78,79,80
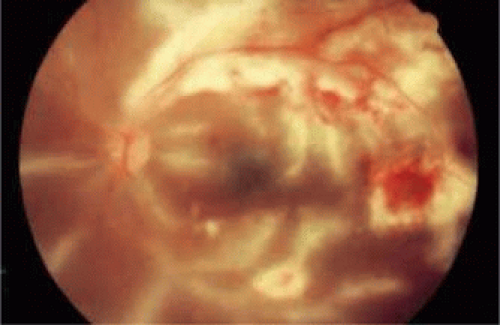 Figure 47.9. Frosted branch angiitis in a patient with AIDS. Cytomegalovirus retinitis is seen superiorly and temporally, as evidenced by retinal hemorrhages and white necrotic retina. Note the frosted branch appearance of the veins inferiorly and adjacent to the disc. |
IDIOPATHIC RETINAL VASCULITIS, ANEURYSMS, AND NEURORETINITIS
Idiopathic retinal vasculitis, aneurysms, and neuroretinitis (IRVAN) syndrome usually affects young, healthy persons and has a slight female predominance.81 There are no systemic abnormalities. Retinal findings include dilatation of the retinal and optic nerve head arterioles, vasculitis, neuroretinitis, and extensive peripheral capillary nonperfusion. Retinal and anterior segment neovascularization, optic nerve head swelling, retinal ischemia, macular edema, neovascular glaucoma, and anterior uveitis can occur.82 This disease can have major sight-threatening consequences. The primary causes of visual loss in this disease are retinal ischemia and neovascular glaucoma.82 Systemic corticosteroids have little effect on the progression of the disease.81 Although other treatments are being investigated, there have been promising results reported if panretinal photocoagulation is done early in the disease course.82
CHORIORETINAL INFLAMMATORY DISEASE
BIRDSHOT CHOROIDOPATHY
Birdshot choroidopathy is a bilateral intraocular inflammation affecting patients in the fifth to eight decades of life, with a slight female predilection. The most common complaints include floaters, progressive loss of vision, photopsias, and nyctalopia. The characteristic fundus findings are multiple depigmented or cream-colored lesions at the level of the retinal pigment epithelium and choroid.
This disease is strongly associated with the presence of human leukocyte antigen (HLA) A29. More recently, the HLA class 1-specific killer cell immunoglobulin-like receptor (KIR) genes have been implicated in the disease pathogenesis.83 Histopathology may show lymphocytic aggregates around retinal vessels and the optic nerve.84
Retinal vascular abnormalities commonly seen in birdshot choroidopathy include narrowing of retinal arterioles, sheathing of retinal vessels and vascular tortuosity.85 Retinal vascular leakage, papillitis, and cystoid macular edema may be seen on fluorescein angiography.86 Fundus autofluorescence shows patchy areas of hypofluorescence that may represent areas of RPE atrophy, which may or may not correspond to the clinically seen cream-colored lesions.87
RADIATION RETINOPATHY
Radiation damage to small retinal blood vessels may result in cotton-wool spots, retinal hemorrhages, microaneurysms, hard exudates, telangiectases, perivascular sheathing, macular edema, and capillary nonperfusion. The retinopathy can be proliferative or nonproliferative.88 Radiation damage can also cause choroidal hypoperfusion.89 Fluorescein angiography shows capillary occlusion and dropout and peripheral ischemia.90 Vitreous cells are rare. Radiation retinopathy usually occurs several years after radiation is administered. Although direct effect on the vascular endothelium is thought to be the primary pathogenic mechanism, there is evidence for immune complex–mediated interactions.91 Vascular endothelial growth factor (VEGF) has also been implicated in its pathogenesis, supported by a recent study that showed the anti-VEGF agent, bevacizumab, to be helpful in reduction of hemorrhage, exudates, and edema associated with radiation retinopathy.92,93
SYSTEMIC VASCULITIS
SYSTEMIC LUPUS ERYTHEMATOSUS
Systemic lupus erythematosus is characterized by autoantibody formation and small vessel occlusions. Ocular manifestations occur in approximately 15% of SLE patients, with retinal vasculitis reported in 5% of these patients.94 The retinopathy of SLE is primarily a diffuse arteriolar occlusive vasculitis,95,96 and there may be microangiopathy with cotton-wool spots and intraretinal hemorrhage (Fig. 47-10).97 Patients with only cotton-wool spots have a favorable prognosis.98 However, vaso-occlusive retinopathy carries a more severe prognosis, with about 50% of patients eventually becoming legally blind.99
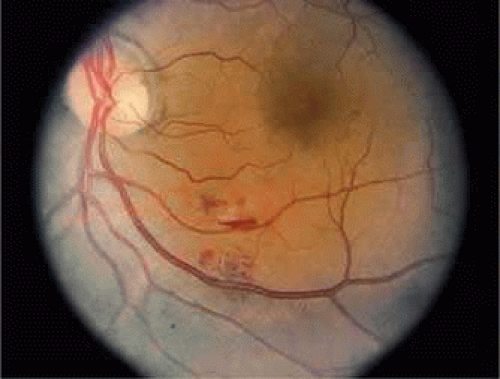 Figure 47.10. Patient with lupus vasculitis. Note the cotton-wool spot adjacent to the inferotemporal artery with a small hemorrhage. |
Retinal manifestations of SLE may also include capillary nonperfusion, venous stasis, and retinal edema. Occasionally, periphlebitis, perivenous sheathing, and branch or central vein occlusion are also found. Fluorescein angiography may show focal leakage from capillaries and arterioles, optic disc leakage, large vessel obstruction, delayed venous filling, microaneurysms, capillary nonperfusion, and neovascularization.
Involvement of the central nervous system has been reported in 73% of patients with severe lupus retinopathy.99 A lupuslike syndrome of multiple branch retinal artery occlusions and neurologic disturbances affecting young women has been described.100 Although these patients do not fulfill the criteria for SLE, their disease suggests immune complex–mediated vasculitis similar to SLE. Patients with idiopathic retinal vasculitis may have positive blood tests for antiphospholipid antibodies, lupus anticoagulant, and antinuclear antibodies without having definitive SLE.
RHEUMATOID ARTHRITIS
Rheumatoid arthritis is a chronic systemic disease with symmetric inflammation of small joints, rheumatoid nodules, and a positive test for serum rheumatoid factor. Systemic vasculitis involving small to medium-sized vessels can occur.103 Retinal vasculitis was found on fluorescein angiography in 18% of patients with definite or classic rheumatoid arthritis, even in the absence of clinical or ophthalmoscopic evidence of retinal vessel inflammation.104 Retinal vasculitis tends to occur during active phases of rheumatoid arthritis, as shown by a positive C-reactive protein test and an elevated erythrocyte sedimentation rate (Fig. 47-11).103,104
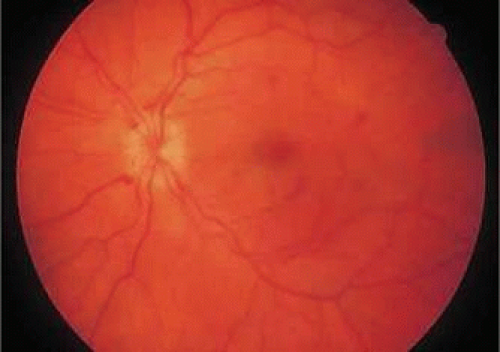 Figure 47.11. Patient with rheumatoid arthritis and retinal vasculitis. Note the optic disc swelling with retinal hemorrhages around the disc and in the periphery. The patient later developed cerebral vasculitis with vertical nystagmus. |
OTHER CONNECTIVE TISSUE DISEASES
Dermatomyositis and relapsing polychondritis have been associated with an ischemic retinopathy that resembles lupus.105,106 Retinopathy associated with dermatomyositis is rare and usually resolves without residual complications. Retinal findings may include cotton-wool spots, intraretinal hemorrhages, and macular edema, suggestive of an occlusive vasculitis process.107 Profound visual loss is caused by macular hemorrhage or edema, which produces central scotomas. Relapsing polychondritis is a rare connective tissue disease with inflammation of the auricular, nasal, and laryngotracheal cartilage.108 Ocular findings include episcleritis, scleritis, proptosis, corneal infiltrates, iridocyclitis, optic neuritis, ischemic optic neuropathy, exudative retinal detachment, chorioretinitis, cotton-wool spots, vascular occlusions, and retinal vasculitis.106
SUSAC SYNDROME
Susac syndrome is an occlusive arteriolar disease characterized by infarcts in the retina, cochlea, and brain, occurring most commonly in young women. Findings include recurrent multifocal branch retinal artery occlusions, sensorineural hearing loss, and neuropsychiatric abnormalities. Retinal arterial wall plaques, yellow-white and usually appearing in midarterial sections, have also been reported.109 Fluorescein angiogram shows occlusion of retinal arterioles in the posterior pole and periphery. Indocyanine green angiography shows an intact choroidal circulation.110 A patient with scleroderma who developed all the features of Susac syndrome has been reported (Fig. 47-12).111 Another patient developed arterioarterial collaterals in the retina.112
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


