Ocular Manifestations of Hematologic Diseases
Ramin Sarrafizadeh
Richard S. Kaiser
Amy S. Noffke
George A. Williams
Disorders of Hemostasis and Coagulation
The ocular manifestations of hematologic diseases are protean. They reflect both the high vascularity and the unique vascular anatomy of the eye. Hematologic diseases include disorders of erythrocytes, leukocytes, and platelets, as well as disorders of coagulation and plasma proteins. These diseases may present with ocular involvement, or ocular manifestations may arise during their course. Ocular findings also may occur as a consequence of treatment of these disorders.
In the past 20 years, substantial advances in the understanding of various hematologic disorders have been made. These advances also have resulted in better pathophysiologic comprehension of ocular manifestations of hematologic disease. This chapter discusses these manifestations, with an emphasis on thrombophilia (hypercoagulable states), coagulopathies, and disorders of the cellular elements of the blood with the exception of the hemoglobinopathies, which are discussed elsewhere in this text.
Disorders of Hemostasis and Coagulation
The normal hemostatic mechanism is an intricately balanced system that regulates the formation and clearance of blood clots precisely.1 A variety of stimuli including traumatic, inflammatory, and metabolic events can stimulate coagulation through the activation of complex enzymatic cascades. The biochemical integrity of these cascades is dependent on the presence and function of multiple factors, some of which are proteases responsible for the conversion of zymogens into other proteases. Other factors act as catalysts or cofactors, enhancing the enzymatic activity of various proteases. The end product of these hemostatic cascades is the formation of polymerized fibrin, which is the major component of clotted blood (Fig. 23.1). The sequential activation of these factors is separated into two distinct, yet interdependent, pathways: the extrinsic and the intrinsic pathways. Although these pathways involve both different activating stimuli and different factors, their end product, activated factor X, is the same. Activated factor X then stimulates the formation of thrombin, which subsequently generates fibrin monomers, which are, in turn, polymerized into fibrin strands by factor XIII. Simultaneously with the activation of the coagulation system, the fibrinolytic system also is activated, resulting in the formation of plasmin, which clears fibrin.
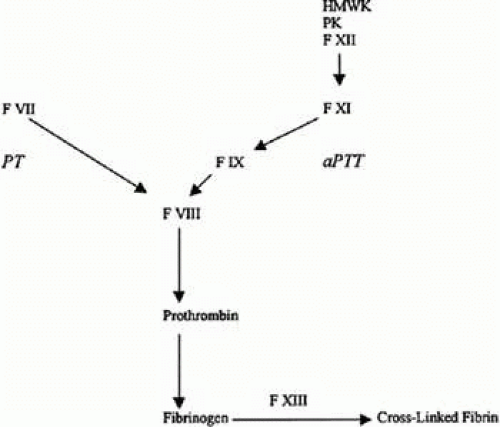 FIGURE 23.1 Coagulation cascade. Prothrombin time (PT) measures the extrinsic pathway and the activated prothrombin time (aPTT) measures the intrinsic pathway. HMWK = high-molecular weight kininogen; PK = prekallikrein. (Adapted from Cecil’s Textbook of Medicine.) |
Within the extrinsic, intrinsic, and fibrinolytic cascades, there are complex autoregulatory mechanisms that tightly control their respective enzymatic activities. The interdependence of these separate pathways is perhaps best shown in disease states in which individual factors are lacking or dysfunctional as a result of either hereditary or acquired disorders. The severe hemorrhagic complications resulting from the absence of a single factor underscore the interdependence of the individual factors and pathways in the maintenance of hemostasis. These deficiencies can result in a spectrum of clinical manifestations ranging from subtle bleeding events to fulminant hemorrhagic crises. This section briefly reviews some of the coagulation disorders, both hereditary and acquired, that have been associated with ocular disease.
Coagulation Factor Deficiencies
The most common inherited coagulation disorders involve X-linked deficiencies of factors VIII (hemophilia A) and IX (hemophilia B), which are necessary for the generation of activated factor X by the intrinsic pathway. These disorders are clinically indistinguishable and are characterized by insidious onset of hemorrhage occurring hours to days after injury. The severity of the bleeding is correlated with the extent of the respective factor’s deficiency. Patients with less than 1% of normal activity have severe hemorrhagic disease, usually presenting with complications at birth. Levels of factor activity from 1% to 5% of normal are considered moderate, and bleeding may not become apparent until the patient begins crawling or walking. Mild hemophilia with levels of from 5% to 25% of normal may not be diagnosed in patients until adolescence or after major trauma or surgery. The classic presentation of hemophilia involves pain and swelling in a weight-bearing joint because of hemarthrosis. Chronic synovitis may result, with long-term disabling sequelae. Intramuscular hematomas are common, and large hematomas may compress vital structures. Central nervous system (CNS) bleeding accounts for 25% of the deaths due to hemophilia and has a mortality rate of 34%.2 Although replacement therapy of the respective deficient factor in hemophilia has greatly improved the prognosis, it also has resulted in other severe problems such as hepatitis and acquired immunodeficiency syndrome (AIDS) from virally contaminated blood products. The advent of recombinant DNA replacement therapy has eliminated these complications.
Deficiencies of other factors are much less common than the hemophilias. Factor XI deficiency is an autosomal-recessive disorder with relatively minor clinical manifestations such as epistaxis, hematuria, and menorrhagia. It usually is detected when bleeding ensues after surgery. The disparity in clinical severity between factor XI deficiency and the hemophilias is explained by the ability of the extrinsic system to bypass factors XII and XI through direct activation of factor IX. Thus, an intact extrinsic pathway can compensate for dysfunction early in the intrinsic pathway.1
Although ocular and periocular hemorrhages may occur, particularly after trauma or surgery, ocular manifestations of hemophilia are primarily neuro-ophthalmologic in nature, resulting from CNS hemorrhage. Pupillary abnormalities, cranial nerve palsies, visual blurring, and papilledema have been described after intracranial bleeding.2,3 Repeated retinal hemorrhages and vitreous hemorrhage have been associated with factor VIII and factor IX deficiency.4,5,6 Hemorrhagic choroidal detachment has been reported following retinal detachment surgery in a patient with spontaneously acquired factor VIII inhibitor.7
Thrombophilia
The hypercoagulable state of thrombophilia is associated with both genetic and acquired defects of hemostasis that result in a predisposition to venous thrombosis through the malfunction of the normal balance between anticoagulant and procoagulant factors. Thrombophilia is a multigenic disorder resulting from both genetic and acquired disorders.8 Genetic risk factors for thrombophilia include factor V Leiden abnormality which causes activated protein C resistance, prothrombin variant 20210a, antithrombin III deficiency, protein C deficiency, and protein S deficiency. Acquired disorders causing thrombophilia include antiphospholipid antibody syndromes, hyperhomocysteinemia, and abnormal lipoprotein(a) levels.
Activated Protein C Resistance/Factor V Leiden
Protein C is a vitamin K-dependent plasma protein that acts as a physiologic anticoagulant. Protein C circulates as an inactive zymogen and is activated by enzymatic cleavage on the surface of endothelial cells by a complex of thrombin and thrombomodulin. Activated protein C is an important endogenous anticoagulant factor that inactivates the procoagulant factors V and VIII, thereby limiting the production of thrombin.9 A hypercoagulable state can result from resistance to the effects of activated protein C as measured by the activated partial thromboplastin time (PTT) assay.10 Activated protein C resistance has been shown to be the result of heterozygosity or homozygosity of a single-point mutation in the factor V gene resulting in a mutant factor V, in which arginine is replaced by glutamine at position 506.11 This is now termed factor V Leiden, which accounts for more than 80% of the cases of activated protein C resistance.12 To date, no other mutations have been associated with activated protein C resistance. Factor V Leiden is the most common genetic predisposition to venous thrombosis. It occurs in 5% to 7% of whites but is rare in non-white populations.8,12,13 It has been reported to be as common as occurring in up to 15% of some European populations.14
The association of activated protein C resistance and factor V Leiden with ocular thrombotic disease has been controversial. Activated protein C resistance was first reported in patients with systemic thrombophilia and central or branch retinal vein occlusions.15,16 However, other reports have failed to show a positive correlation between factor V Leiden and retinal vascular occlusions.17,18 Some reports have been suggestive of a correlation between factor V Leiden and retinal vein occlusions.19 Patients with central retinal vein occlusions (CRVOs) were found to have a higher prevalence of factor V Leiden mutations compared with that of the general populations.20,21,22 Heterozygosity for the mutant allele has been associated with a 5-fold to 10-fold increased risk of venous thrombosis and homozygosity with a 50- to 100-fold risk.23 Studies of patients with Behçet’s disease found that those with ocular and systemic thromboembolic disease had a higher than normal incidence of factor V Leiden mutation.24 Overall, because of the high prevalence of factor V Leiden mutations in the general population and the small number of people with the mutation in whom venous thromboembolism develops, it seems clear that factor V Leiden carriers have thrombosis develop along with other risk factors.25 In younger patients, factor V Leiden may be more prevalent in those who develop a retinal vein occlusion.26,27,28,29 Additionally, factor V Leiden alone probably is not an important risk factor for arterial disease; however, in the presence of other risk factors, such as smoking, atherosclerosis, and other genetic defects, it may be an associated arterial risk factor.14
Prothrombin Mutations
A genetic risk factor that predisposes to venous thrombosis has been identified in the gene for prothrombin involving a mutation of nucleotide position 20210a. This mutation occurs in 1% to 2% of healthy individuals, 6% of individuals with the first onset of thromboembolic disease, and up to 18% of patients with unexplained familial thrombophilia. It is the second most common inherited abnormality, next to factor V Leiden, which leads to an increased risk of thrombosis. The mechanism by which prothrombin variant 20210a causes thrombophilia is unknown. Screening for this defect relies on DNA analysis. There is a strong interaction between this defect and other genetic risk factors, particularly the factor V Leiden mutation.8,12,13 Some reports suggest an association between prothrombin variant 20210a and ocular vascular disease. However, one report failed to confirm an association between prothrombin variant 20210a and central or branch retinal vein occlusions.20
Protein C Deficiency
Protein C deficiency has been associated with more than 160 mutations.30 Although homozygous deficiencies of protein C tend to result in severe thrombotic disease, heterozygous deficiencies and acquired defects result in milder thrombotic tendencies. Heterozygosity for protein C deficiency occurs in 0.3% of healthy individuals according to blood donor statistics.31 It is estimated to occur in approximately 6% of patients with inherited thrombophilia. A heterozygous deficiency of protein C confers a sevenfold increase in the risk of venous thromboembolic disease, and the risk increases with lower protein C levels.
Protein C is a vitamin K-dependent protein. Initiation of anticoagulation therapy with vitamin K antagonists may cause thrombosis in patients with protein C deficiency, which has been implicated in retinal vein occlusions and amaurosis fugax.32,33,34,35,36 Deficiency also has been associated with vitreous hemorrhages and hemorrhagic retinal and choroidal detachment in neonates.37 Retinal arteriolar occlusions have been reported in a patient with protein C deficiency.38
Protein S Deficiency
Protein S is a nonenzymatic cofactor necessary for the anticoagulant activity of activated protein C. Protein S by itself also has anticoagulant activity by forming a complex with C4b binding protein, a regulatory protein of the complement system. Protein S deficiency is inherited as an autosomal-dominant trait. Unlike protein C deficiency or antithrombin III deficiency, heterozygous protein S deficiency is not as strong a risk factor for thrombosis. Nevertheless, central retinal vein occlusion secondary to protein S deficiency has been reported.40,41 Retinal artery occlusion has been described with protein S deficiency.32,39,42 Figure 23.2 shows a pregnant woman with protein S deficiency and a branch retinal artery occlusion.
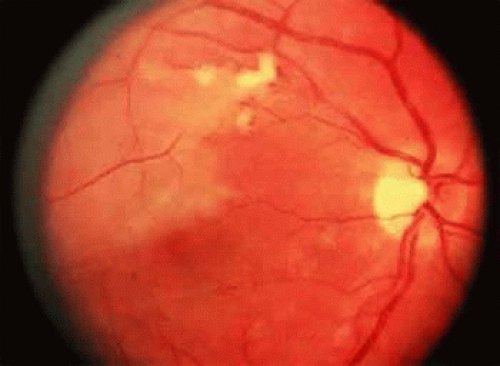 FIGURE 23.2 A 32-year-old woman during her third trimester of pregnancy with a branch retinal artery occlusion. She was found to have protein S deficiency. |
Antithrombin III Deficiency
Antithrombin III is an endogenous inhibitor of thrombin and other pro-coagulant factors and thus is a major coagulation inhibitor. Elevated systemic thrombin-antithrombin III complexes indicate higher thrombin formation and, thus, a hypercoagulable state in the systemic circulation. Studies have shown elevated thrombin-antithrombin III complexes in patients with vein occlusions compared with those of the general population.43
Antithrombin III deficiency is inherited as an autosomal-dominant defect with heterozygosity conferring an increased risk of venous thrombosis. The prevalence of antithrombin III deficiency is increased in patients with inherited thrombophilia (4% vs. 0.02%). More than 70 specific mutations have been identified. Antithrombin III deficiency has been associated with central retinal vein occlusion and cilioretinal artery occlusion.34
Antiphospholipid Syndromes and Lupus Anticoagulant
The antiphospholipid antibody syndrome is a clinical syndrome of venous and arterial thrombotic events. Antiphospholipid antibodies consist of two different antibodies: (1) the lupus anticoagulant and (2) the anticardiolipin antibody. Although the two are similar, there are distinct clinical, laboratory, and biochemical differences. The anticardiolipin antibody antiphospholipid syndrome is more common than the lupus anticoagulant syndrome. Both of these antibodies are associated with thrombosis in connective tissue diseases such as lupus and autoimmune diseases, malignancy, AIDS, and multiple drugs.44 However, some cases of lupus anticoagulant and anticardiolipin antibody are found in otherwise healthy patients. These people are considered to have primary antiphospholipid antibody syndrome. Both syndromes may be associated with arterial and venous thrombosis, fetal loss, and thrombocytopenia. However, the anticardiolipin syndrome often is associated with both arterial and venous thrombosis, including premature coronary and cerebrovascular disease and retinal vascular disease.45 The lupus anticoagulant, although sometimes associated with arterial disease, is more often associated with venous thrombosis.44
Lupus anticoagulant is an immunoglobulin G antibody to anionic phospholipids that is related to anticardiolipin and the biologic false-positive test result for syphilis. This antibody is deemed anticoagulant because of its in vitro effects on results of coagulation tests such as the PTT test. Despite these in vitro effects, lupus anticoagulant is characterized clinically by ocular and systemic thrombotic events.44,46 The mechanism of clinical thrombosis is uncertain, but it probably involves interference with normal coagulation mechanisms by the antibodies. It is hypothesized that the antibodies act on components of platelet membranes and vascular endothelium as well as on natural anticoagulant proteins such as protein C, protein S, and antithrombin III.45
The ocular manifestations of antiphospholipid antibodies include retinal venous and arterial occlusions, amaurosis fugax, diplopia, and visual field loss.47,48,49,50,51,52,53,54 Extensive vaso-occlusion, neovascularization, and vitreous hemorrhage may occur (Fig. 23.3). Treatment consists of photocoagulation for the neovascularization and systemic anticoagulation and immunosuppression. Vitrectomy may be required for vitreous hemorrhage. The role of systemic treatment of lupus anticoagulant in the management of ocular disease is unclear. Some investigators suggest that systemic anticoagulation be started promptly.54 The optimal duration of anticoagulation and whether antiplatelet therapy should be used are not known, however. The optimal use of corticosteroids or other immunosuppressive drugs such as cyclophosphamide and azathioprine also is unknown. Two reports have found an association with the antiphospholipid syndrome and some additional retinal conditions. One report also has associated the primary antiphospholipid syndrome with central serous chorioretinopathy.55 In addition, lupus anticoagulant positivity could represent an additional risk factor for diabetic retinopathy.56
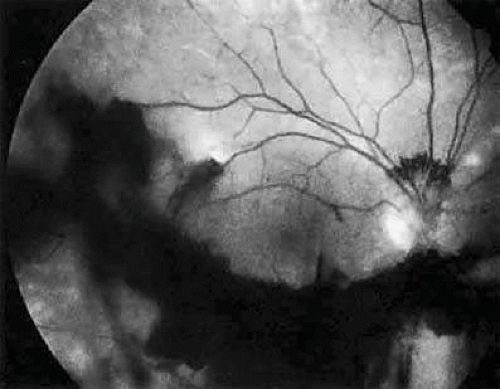 FIGURE 23.3 Retinopathy associated with lupus anticoagulant with disc neovascularization and vitreous hemorrhage. Source: Kleiner RC, Najarian LV, Schatten S et al: Vaso-occlusive retinopathy associated with antiphospholipid antibodies [lupus anticoagulant retinopathy]. Ophthalmology 96:898, 1989. |
Lupus anticoagulant should be suspected in all cases of occlusive retinopathy without obvious causes, especially in young adults. Although the diagnosis of lupus anticoagulant is suggested by prolongation of the PTT, this test is not reliable as a screening assay. The Russell viper venom time and the tissue thromboplastin inhibition test have been advocated as sensitive and relatively specific tests for detection of lupus anticoagulant. Anticardiolipin antibodies can be measured by enzyme-linked immunosorbent assay, which can be more sensitive.57,58,59
Hyperhomocysteinemia
The term hyperhomocysteinemia describes a variety of disorders that cause elevated plasma levels of homocysteine, homocystine, and their metabolites. Homocysteine is an amino acid formed during the metabolism of methionine. Vitamins B12 and B6 and folate serve as cofactors in the metabolism of methionine (Fig. 23.4).
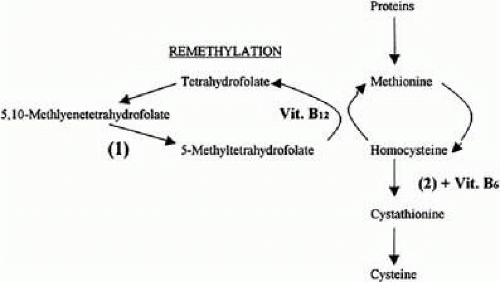 FIGURE 23.4 Homocysteine pathway. Numbers indicate principal enzymes involved in hyperhomocysteinemia. (1) 5,10-methylenetretrahydrofolate reductase and (2) cystathionine-beta-synthase. Source: Adapted from Cecil’s Textbook of Medicine. |
Hyperhomocysteinemia is an independent risk factor for thrombophilia and usually is caused by genetic enzymatic defects in homocysteine metabolism or nutritional deficiencies in vitamin cofactors. Congenital homocystinuria is a rare homozygous deficiency associated with marked hyperhomocysteinemia resulting in premature vascular disease. Ocular manifestations of congenital homocysteinemia include dislocation of the lens and retinal vascular occlusions. Heterozygous deficiencies of cystathionine B-synthase or methylenetetrahydrofolate reductase are relatively common. Nutritional deficiencies of vitamin cofactors such as folate, vitamin B12, and vitamin B6 contribute to approximately two-thirds of all cases of hyperhomocysteinemia. Vitamin supplementation can normalize high homocysteine concentrations.
Overall, mild hyperhomocysteinemia occurs in 5% to 7% of the general population.60,61 Some studies have estimated that 10% of the risk of coronary artery disease in the general population is attributable to hyperhomocysteinemia.62 Ophthalmic studies have shown an increased incidence of hyperhomocysteinemia in young adults with retinal artery and vein occlusions.63,64,65 One study found that hyperhomocysteinemia is a risk factor for central retinal vein occlusions and was found in a high percentage of bilateral CRVOs (55%) and ischemic CRVOs (30%) and was associated with severe vision loss.66 An association between hyperhomocysteinemia and nonarteritic anterior ischemic neuropathy67,68 and neovascular age-related macular degeneration has also been reported.69,70
Fibrinolysis
Fibrinolysis is responsible for the degradation of blood clots via plasmin. Plasmin activity is controlled by a balance between plasminogen activators and plasminogen activator inhibitors. Inhibition of plasmin activity (hypofibrinolysis) is a cause of thrombophilia. Hypofibrinolysis may occur from genetic mutations in plasminogen activator inhibitor activity or plasminogen activity.13 Some have speculated that hypercoagulability and fibrin deposition trigger fibrin(ogen)olysis, which can be hampered to some extent by elevated levels of lipoprotein(a).71 Thus, high levels of lipoprotein(a) interfere with plasminogen activation. Dysfunctional fibrinolysis has been correlated with central vein occlusion.20,71,72
Thrombophilia Screening
Thrombophilia is a hypercoaguable state resulting from genetic or acquired abnormalities of the hemostatic mechanism that predispose to thrombosis. Multiple mechanisms contribute to thrombophilia, making it a multigenic disorder. The detection and treatment of thrombophilia are directed toward preventing life-threatening vascular disease such as deep vein thrombosis, pulmonary embolism, myocardial infarction, and cerebral vascular disease in the patient or potentially affected family members. The use of systemic anticoagulation to treat this condition remains controversial.73 There are no data to guide the ophthalmologist on the role of anticoagulation in the treatment of thrombophilia and related ocular complications.
In the laboratory, the main abnormalities to search for are hyperhomocysteinemia, factor V Leiden, prothrombin variant (gene 20210a), antiphospholipid antibodies, antithrombin III, protein C and protein S deficiencies, and hypofibrinolysis (Table 23.1).
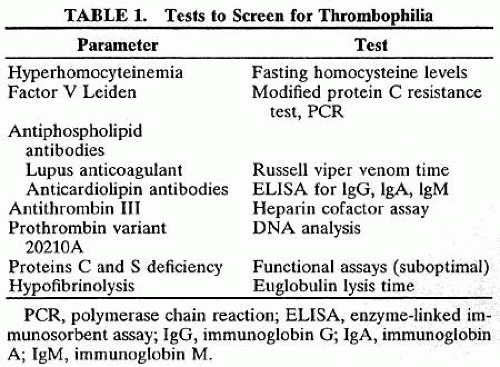 |
Detecting hyperhomocysteinemia has important clinical implications. These patients can be treated with vitamin supplementation instead of long-term anticoagulant treatment. The diagnosis of hyperhomocysteinemia can be made by measuring blood levels of homocyst(e)ine during fasting or by giving an oral dose of methionine with subsequent measurement of homocyst(e)ine blood concentrations.61
Screening tests for activated protein C resistance (factor V Leiden mutation) are easy to perform and have a high sensitivity and specificity (almost 100%). Multiple factors make screening for factor V Leiden using a PTT-based test unreliable and thus impractical.17 Screening for activated protein C with newer molecular testing is highly sensitive and specific, making molecular testing the definitive diagnostic method.8,12,13 The “modified activated protein C resistance test” is based on coagulation assays that dilute the patient’s plasma sample with factor V-deficient plasma. This enables identification of patients with or without activated protein C resistance. Furthermore, the modified activated protein C resistance test has been shown to be more sensitive and specific in identifying protein C resistance in children.74 Molecular biology (polymerase chain reaction) is now only needed to confirm the factor V abnormality.14
Assays to look for prothrombin gene variant 20210a are not commonly performed. Screening for this defect relies on DNA analysis and is impractical. Information on testing positive for this genetic variant would be most beneficial in an individual with other genetic factors, particularly the factor V Leiden mutation.
Antiphospholipid antibodies can be detected by coagulation or immunologic assays. Lupus anticoagulant assays do not actually measure a titer of antibody but are functional tests. The in vitro detection methods for lupus anticoagulant appear to have no relevance to its in vivo actions. Lupus anticoagulants prolong clotting times in vitro because they agglutinate phospholipids in the plasma, thereby preventing their participation as cofactors in coagulation steps. When a patient is suspected of having lupus anticoagulant, performing a PTT test is not adequate. A more definitive test is the Russell viper venom time.44 Similarly, a PTT test usually is not prolonged in patients with anticardiolipin antibodies, and thus definitive tests such as enzyme-linked immunosorbent assays for immunoglobulin G (IgG), immunoglobulin A (IgA), and immunoglobulin M (IgM) anticardiolipin antibodies should be performed. Similarly, screening for anticardiolipin antibodies requires coagulation and immunologic assays.14
Antithrombin III deficiency is simple to detect in most coagulation laboratories using a standardized heparin cofactor assay.14 There are few variations in assay levels between patients of different ages or sex, and therefore a narrow reference range is used to determine antithrombin III abnormalities. The genetic defects of antithrombin III deficiency have been identified but do not need to be tested to identify the defect.
Screening for protein C deficiency uses clotting or amidolytic assays. There is no easy functional assay for protein C deficiency. Multiple factors may affect assay results, including age, sex, and hormonal therapy, thus causing overlap between normal subjects and heterozygotes.12,13 Molecular characterization is possible, but the large number of defects that exist makes this an impractical screening method.14
Screening for protein S deficiency relies on functional assays that are suboptimal because of high percentages of false-positive and false-negative results. Currently, most laboratories use enzyme-linked immunosorbent assays to measure free and total protein S antigen. There is no proven value of functional assays measuring protein S activity.14 Thus, the diagnosis of protein S deficiency remains difficult.
Assays to look for hypofibrinolysis (i.e., reduced level of tissue plasminogen activator or increased levels of plasminogen activator inhibitor) are not performed. There is a large variation of results, and baseline fibrinolytic value does not predict the occurrence of future thrombosis.
Defining which patients to screen for thrombophilia is controversial. Some general guidelines include selecting patients who have a family history of thromboembolic disease, recurrent thrombosis, an early age of onset, both arterial and venous thrombosis, or those who have thrombosis and fetal losses. It is important to rule out common acquired causes of thrombophilia such as cancer before extensive laboratory investigation. Additionally, tests should only be performed if they are going to change the treatment of the patient or if they potentially have consequences for the patient’s family. Finding an abnormality rarely influences the management of an acute event. Moreover, patients who are anticoagulated, pregnant, or taking oral contraceptives have altered thrombophilia test results. These factors must be accounted for in patient screening and testing.
The primary reason to investigate a patient and their family is to give specific prophylactic and therapeutic recommendations for a sustained risk for thrombosis (Table 23.2). For example, a symptomatic patient with multiple thrombotic events may be treated with anticoagulation for a variable duration depending on the number of risk factors detected and the clinical circumstance. Conversely, patients who are diagnosed with hyperhomocysteinemia can be treated with vitamin supplementation instead of long-term anticoagulant treatment. Another scenario in which the subject would benefit from screening is an asymptomatic family member who is found to be a carrier of a thrombophilia genetic defect. Depending on the number of risk factors, the patient may receive prophylaxis anticoagulation during high-risk situations such as pregnancy, surgery, or immobilization. A patient also may benefit from the knowledge that he or she is more predisposed to be hypercoaguable by changing lifestyle issues such as stopping smoking, avoiding oral contraceptives, and tightly controlling hypertension.
 |
There are no data to support thrombophilia screening for the general population. A combined clinical and laboratory approach, taking into account the history of the patient and the patient’s family and the sensitivity and specificity of the tests, should decide which tests are most appropriate for a patient.14
Thrombophilia Treatment and Anticoagulant Therapy
Bleeding disorders associated with anticoagulant therapy probably are more common than inherited or acquired coagulation deficiencies.75 Anticoagulants are used therapeutically to prevent or limit thrombosis in a variety of conditions such as pulmonary embolism, ischemic cerebrovascular disease, cardiac disease, and postoperative immobility.
Heparin is used for acute anticoagulation, and the coumarin group of anticoagulants is used for chronic therapy. Heparin is a naturally occurring mucopolysaccharide that enhances the activity of antithrombin III, the major endogenous anticoagulant factor. In the presence of heparin, antithrombin III more effectively neutralizes thrombin and other activated coagulant factors, thereby slowing coagulation. Recently, low-molecular-weight heparins have established themselves as an important class of antithrombotic compounds. They produce a more predictable anticoagulant response and cause less bleeding than unfractionated heparin. The low-molecular-weight heparins are being used with greater frequency because of these advantages.76 The coumarin anticoagulants interfere with vitamin K metabolism, thereby affecting production of thrombin and factors VII, IX, and X and thus slowing coagulation.1 There is a need for more potent anticoagulant drugs that are able to inactivate thrombin bound to fibrin, which may be an important trigger for clot extension at the sites of vascular injury.76 New drugs may enable more effective treatment of thrombotic diseases.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


