Chapter 156 Primary Vitreoretinal Lymphoma
Introduction
PCNSL is a variant of extranodal non-Hodgkin lymphoma (NHL) that is predominantly a high-grade B-cell malignancy associated with a median survival ranging from 1–8 years depending on factors such as age and Karnofsky performance status.1 It originates in the brain parenchyma, spinal cord, leptomeninges, and eyes.2–4 Formerly used descriptors such as “reticulum cell sarcoma” and “microgliomatosis” are no longer preferred as both misleadingly imply that the lymphoma arises from transformed reticulum or microglial cells.5,6
Epidemiology
PCNSL accounts for 1–2% of all cases of lymphoma and approximately 3–5% of primary tumors of the central nervous system.7–9 From 1973–1997, the incidence of PCNSL increased threefold, partially owing to the corresponding rise in individuals with human immunodeficiency virus (HIV) infection and possibly due to improved diagnosis and/or detection leading to more cases being ascertained.7 Rates have since stabilized with an age-adjusted incidence of PCNSL in the USA of 4.8 per million population reported in 2002.7 PCNSL is therefore a rare disease affecting fewer than 2000 individuals annually in the USA.4
While PVRL is frequently seen in the setting of PCNSL, the exact incidence is unknown. Between 1999 and 2002, approximately 100 new cases of PVRL were reported in the USA.10 The association between PVRL and PCNSL is variable, with CNS disease manifesting prior to, following, or occurring simultaneously with ocular presentation. Approximately 25% of patients with PCNSL will have concomitant PVRL.3 In contrast, 56–85% of individuals with PVRL will ultimately develop central nervous system involvement.11–14 Among immunocompetent individuals, the peak incidence of PVRL occurs between ages 50 and 70 years. In the immunocompromised population, PVRL occurs in younger individuals.15–17 There are no known racial or ethnic associations. In a large, multicenter retrospective review of 221 patients with PCNSL with intraocular involvement, a slight female predominance (57%) was observed.18
Etiology and pathogenesis
PCNSL is believed to originate from late-germinal center or post-germinal center lymphoid cells, however the neurotropic mechanism by which these cells localize to the CNS remains uncertain.19 It has been hypothesized that the trafficking of lymphoma cells from the brain to the eye and vice versa involves either invasion of the optic nerve, seeding through shared venous drainage of the brain and eye, or common integrin expression of both organs.4 While there are no known risk factors for the development of PCNSL in immunocompetent individuals, immunosuppression secondary to HIV, congenital immunodeficiency, and iatrogenic immunosuppression are risk factors.20 PCNSL develops in as many as 6% of patients with acquired immune deficiency syndrome (AIDS).21,22 Epstein–Barr virus infection of B-lymphocytes in the absence of T-suppressor lymphocytes results in uncontrolled lymphocytic proliferation. Rare cases of PVRL may be secondary to human T-cell lymphotropic virus type 1 (HTLV-1) infection.23
Clinical findings
Ophthalmic findings
While individuals may be asymptomatic, more than half present with painless, decreased visual acuity or floaters.13,24,25 Of those who are asymptomatic, many are diagnosed when ophthalmic screening examination is performed for individuals with known PCNSL. The clinical findings are bilateral in 80% of cases, but are frequently asymmetric.11
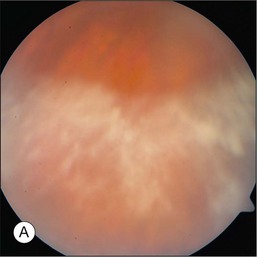
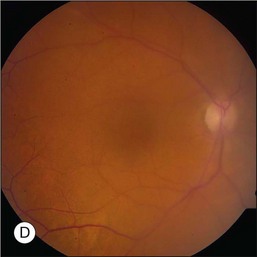
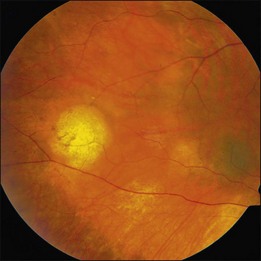
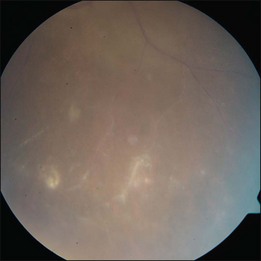
The hallmark of PVRL is the presence of fine vitreous cells or clumps of cells and sub-retinal pigment epithelium (RPE) deposits comprised of aggregated lymphoma cells (Fig. 156.1).26–28 When present, these lesions are considered pathognomonic.28 Anterior segment findings including keratic precipitates, iris nodules, aqueous cells, and flare are frequently encountered but are nonspecific.17,25 On fundus examination, focal, multifocal, or diffuse choroidal, retinal, or chorioretinal infiltrates can be seen in the presence or absence of vitreous cells (Fig. 156.2).26,29 Other less commonly reported findings include: perivasculitis (Fig. 156.3),13 retinal artery occlusion,30 exudative retinal detachment,31 multifocal “punched-out” lesions at the level of the RPE,32 and optic atrophy.33
Central nervous system findings
PCNSL is an aggressive malignancy and the diagnosis is generally established within several months of the onset of symptoms. This is in sharp contrast to PVRL where the diagnosis is often delayed, by as long as 2 years according to one tertiary care center, secondary to being misdiagnosed as posterior uveitis or other masquerading diseases.25 In PCNSL, personality changes are a common presenting symptom as the frontal lobe is the most frequent region of brain parenchymal involvement. Seizures are a rare feature of this disease.
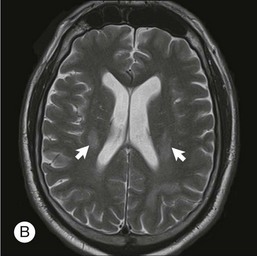
The lesions in PCNSL tend to be periventricular in location thus allowing access to the cerebrospinal fluid (CSF) and leptomeninges. Leptomeningeal involvement occurs in approximately 40% of cases.34 Rarely, PCNSL limited to the spinal cord is observed.35 The lesions can be multifocal, particularly in immunocompromised individuals.
Diagnosis
In the absence of prior CNS disease, the diagnosis of PVRL is based upon histopathological and cytological features. Several techniques exist for biopsy to establish the diagnosis including vitreous biopsy, retinal biopsy, and subretinal biopsy. Most commonly, diagnostic 23-gauge pars plana vitrectomy is performed. Proper surgical techniques and handling are critical as aspirates are generally of low cellularity and fragile lymphoma cells are prone to lysis during sample collection. Techniques vary by center and expertise; however, it is generally recommended that an undiluted vitreous sample of approximately 1–2 mL be collected prior to the start of the saline infusion during vitrectomy.20,36 Following collection of the first sample, the infusion fluid is started, and a second diluted vitreous specimen using gentle vitreous cutting can be collected in a separate syringe.37 Some centers also submit the vitreous cassette as a third sample.38 It is recommended that samples be delivered to the laboratory, without fixative, within one hour of surgery.36,39 Repeated vitreous biopsies are commonly performed in order to establish the diagnosis. In a study of 20 patients with PVRL, three eyes required more than one vitreous biopsy before the diagnosis was confirmed.40 More recently, there has been an interest in using 25-gauge sutureless vitrectomy for diagnostic purposes and for improved patient comfort and decreased operative times. This technique has been used with success in some centers.38
When chorioretinal or subretinal lesions are present, a retinal or subretinal biopsy may be performed. A subretinal biopsy technique using a standard three-port pars plana vitrectomy approach has been described.41 An initial core vitrectomy is performed allowing access to the subretinal infiltrate. Vitreous separation is then induced and thorough vitrectomy is performed over the biopsy site. An incision in the overlying retina just large enough to allow entrance of the vitreous cutter is then made. Suction tubing is then inserted through the retinectomy incision and with gentle cutting action, several samples are obtained. Subretinal aspirates should be placed in a mild cytofixative, such as herpes-glutamic acid buffer mediated organic solvent protection effect (HOPE) fixation or CytoLyt® (Cytyc Corporation).36 In a series of 84 patients who underwent pars plana vitrectomy, additional chorioretinal biopsy with extended immunohistochemistry and polymerase chain reaction (PCR) gene rearrangement studies were performed in three patients after vitrectomy was nondiagnostic. This technique yielded a definitive diagnosis of PVRL in each case.42
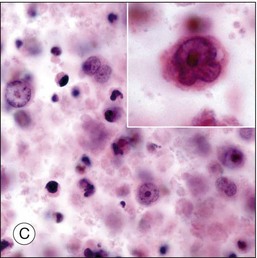
Although diagnostic techniques vary depending upon local expertise, cytology remains the first-line for diagnosis. Access to a pathologist or cytopathologist with experience in ophthalmic diagnosis is essential. The majority of cases of PVRL are diffuse large B-cell lymphomas. In a multicenter retrospective review of 221 individuals with histologically diagnosed PCNSL with ocular involvement, the subtype was large B-cell in 73%, T-cell in 2%, and not specified in 25%.18 The cytologic features are characteristic. PVRL cells are typically 2–4 times larger than normal lymphocytes, are pleomorphic, and have scant cytoplasm.43 The nuclei may be round, oval, or indented, with conspicuous nuclear membranes, occasional fingerlike protrusions, and multiple, prominent, eccentrically located nucleoli. Mitoses are frequently observed.24 With the use of electron microscopy, intranuclear inclusions, cytoplasmic crystalloids, pseudopodal extensions of the cytoplasm, cytosomes, and autophagic vacuoles can be identified.44
As vitreous biopsy is not diagnostic in all cases, supplemental techniques can be helpful in confirming the diagnosis of PVRL. Immunohistochemistry is useful for identifying markers for leukocytes (CD45), B-cells (CD20, CD79a, PAX-5), T-cells (CD45RO), and macrophages (CD68).36 Additionally, clonality can be established with the use of antibodies directed against κ and λ light chains.14,43,45 Flow cytometry provides a means of quantitatively assessing the proportion of cells in a given sample that demonstrate these immunohistochemical markers. PCR gene rearrangement studies can detect monoclonality of the heavy chain variable (V), diversity (D), and joining (J) immunoglobulin gene segments and are therefore helpful in establishing the diagnosis, however most vitreous biopsy samples are inadequate for PCR.46,47 Evaluation for gene rearrangement by PCR is most successful in tissue biopsy specimens in which DNA has been isolated by laser capture microdissection.46,48 Measurement of IL-6 and IL-10 in either aqueous or vitreous fluid can also facilitate diagnosis, although an elevated IL-10/IL-6 ratio is not specific for PVRL.49 Measurement of these cytokines has no role in the routine evaluation of suspected PVRL.
Central nervous system involvement
Due to the high correlation between PVRL and PCNSL, all patients diagnosed with ophthalmic disease should be evaluated by an experienced neuro-oncologist. This evaluation should include neuroimaging and CSF studies. Gadolinium-enhanced MRI of the brain is the imaging modality of choice in evaluating individuals with suspected PCNSL. High-volume (>10 cc) lumbar puncture for protein, glucose, cytology, and flow cytometry is particularly important as leptomeningeal involvement is present in up to 40% of individuals with PCNSL.34 CSF can demonstrate lymphocytic pleocytosis, elevated protein concentration, and low or normal glucose levels. Malignant cells in the CSF are diagnostic for PCNSL. Flow cytometry, however, is the most sensitive and specific marker of CNS lymphoma.50 Additional diagnostic procedures should include CT scans of the chest, abdomen, and pelvis; testicular ultrasound in elderly men; and HIV testing.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


