Sarcoidosis is a multisystem granulomatous disorder that most commonly affects the lungs, skin, lymph nodes, eyes, central nervous system (CNS), reticuloendothelial system, the heart, and bones.
Epidemiology and Etiology
 Sarcoidosis occurs most commonly in African Americans and Caucasians of Northern European descent.
Sarcoidosis occurs most commonly in African Americans and Caucasians of Northern European descent.
 The incidence and prevalence varies among different geographic regions and ethnic groups:
The incidence and prevalence varies among different geographic regions and ethnic groups:
 In the United States, the prevalence among African Americans is 35 to 82 in 100,000 persons, while in Caucasians it is 8 to 11 in 100,000 persons.
In the United States, the prevalence among African Americans is 35 to 82 in 100,000 persons, while in Caucasians it is 8 to 11 in 100,000 persons.
 In Scandinavia, the prevalence is 64 in 100,000 persons.
In Scandinavia, the prevalence is 64 in 100,000 persons.
 Most common organs involved are the lungs (90% to 95%), followed by skin (15% to 20%), lymph nodes (15% to 40%) and eyes (12% to 20%).
Most common organs involved are the lungs (90% to 95%), followed by skin (15% to 20%), lymph nodes (15% to 40%) and eyes (12% to 20%).
 Systemic sarcoidosis typically affects young adults, while ocular sarcoidosis appears to have a bimodal presentation.
Systemic sarcoidosis typically affects young adults, while ocular sarcoidosis appears to have a bimodal presentation.
 Ophthalmic involvement occurs in up to 50% of patients with systemic sarcoidosis, but most series suggest the rate is around 25%.
Ophthalmic involvement occurs in up to 50% of patients with systemic sarcoidosis, but most series suggest the rate is around 25%.
 Ocular involvement is more common among women compared to men, and in African Americans compared to Caucasians.
Ocular involvement is more common among women compared to men, and in African Americans compared to Caucasians.
 Patients typically have a bilateral (98%), granulomatous anterior uveitis. This is the presenting sign of sarcoidosis in 10% to 20% of patients.
Patients typically have a bilateral (98%), granulomatous anterior uveitis. This is the presenting sign of sarcoidosis in 10% to 20% of patients.
 The most common intraocular involvement is anterior uveitis (two-thirds of patients).
The most common intraocular involvement is anterior uveitis (two-thirds of patients).
 The most common site of extraocular involvement is the lacrimal gland.
The most common site of extraocular involvement is the lacrimal gland.
 Although the etiology is unknown, sarcoidosis is believed to be immune-mediated. Both a genetic predisposition (familial aggregation, HLA-B8, HLA-DRB1) and environmental factors (environmental allergens and infectious agents) have been suggested.
Although the etiology is unknown, sarcoidosis is believed to be immune-mediated. Both a genetic predisposition (familial aggregation, HLA-B8, HLA-DRB1) and environmental factors (environmental allergens and infectious agents) have been suggested.
Symptoms
 Blurry vision, floaters
Blurry vision, floaters
 Redness, photophobia, ocular discomfort
Redness, photophobia, ocular discomfort
 Respiratory symptoms (shortness of breath) and constitutional symptoms (fever, fatigue, night sweats, and weight loss)
Respiratory symptoms (shortness of breath) and constitutional symptoms (fever, fatigue, night sweats, and weight loss)
Signs (Figs. 7-1 to 7-7)
 In 2006, the 1st International Workshop on Ocular Sarcoidosis developed criteria for the diagnosis of ocular sarcoidosis, which include seven ocular signs considered suggestive of ocular sarcoidosis (Table 7-1).
In 2006, the 1st International Workshop on Ocular Sarcoidosis developed criteria for the diagnosis of ocular sarcoidosis, which include seven ocular signs considered suggestive of ocular sarcoidosis (Table 7-1).
 Mutton fat (granulomatous) keratic precipitates (KPs), anterior chamber cells and flare, and iris or angle nodules (granulomas). Busacca nodules are in the iris stroma and Koeppe nodules are on the pupillary margin.
Mutton fat (granulomatous) keratic precipitates (KPs), anterior chamber cells and flare, and iris or angle nodules (granulomas). Busacca nodules are in the iris stroma and Koeppe nodules are on the pupillary margin.
![]() The KPs may also be nongranulomatous.
The KPs may also be nongranulomatous.
 Nodules on the trabecular meshwork and/or tent-shaped peripheral anterior synechiae
Nodules on the trabecular meshwork and/or tent-shaped peripheral anterior synechiae
 Vitreous cells, vitreous haze, snowballs sometimes with a “string of pearls” appearance, and snowbanks.
Vitreous cells, vitreous haze, snowballs sometimes with a “string of pearls” appearance, and snowbanks.
 Multifocal, cream-colored, peripheral chorioretinal lesions (Dalen-Fuchs nodules), either active or atrophic
Multifocal, cream-colored, peripheral chorioretinal lesions (Dalen-Fuchs nodules), either active or atrophic
 Retinal vasculitis, perivenous sheathing (candle wax drippings, also known as taches de bougie), and/or a retinal macroaneurysm in an inflammed eye.
Retinal vasculitis, perivenous sheathing (candle wax drippings, also known as taches de bougie), and/or a retinal macroaneurysm in an inflammed eye.
 Optic disk or choroidal granulomas
Optic disk or choroidal granulomas
 Bilaterality
Bilaterality
 Other ophthalmic findings include:
Other ophthalmic findings include:
 Lacrimal gland enlargement
Lacrimal gland enlargement
 Subconjunctival granulomas
Subconjunctival granulomas
 Optic neuritis
Optic neuritis
 Heerfordt’s syndrome (uveo-parotid fever): anterior uveitis, parotid gland enlargement, facial palsy, and fever
Heerfordt’s syndrome (uveo-parotid fever): anterior uveitis, parotid gland enlargement, facial palsy, and fever
 Retinal neovascularization, sometimes with a sea fan appearance.
Retinal neovascularization, sometimes with a sea fan appearance.
Differential Diagnosis
 The major systemic signs include:
The major systemic signs include:
 Respiratory dysfunction
Respiratory dysfunction
 Skin macules and papules, including erythema nodosum and lupus pernio
Skin macules and papules, including erythema nodosum and lupus pernio
 Cranial nerve palsies
Cranial nerve palsies
 Ataxia and cognitive dysfunction
Ataxia and cognitive dysfunction
 Cardiomyopathy and arrhythmias
Cardiomyopathy and arrhythmias
 Vogt-Koyanagi-Harada (VKH) syndrome
Vogt-Koyanagi-Harada (VKH) syndrome
 Sympathetic ophthalmia
Sympathetic ophthalmia
 Multifocal choroiditis
Multifocal choroiditis
 Primary intraocular lymphoma (retinal lymphoma)
Primary intraocular lymphoma (retinal lymphoma)
 Tuberculosis
Tuberculosis
 Syphilis
Syphilis
 Lyme disease
Lyme disease
 Blau syndrome and juvenile idiopathic arthritis in children
Blau syndrome and juvenile idiopathic arthritis in children
Diagnostic Evaluation
 Definitive diagnosis requires tissue biopsy showing noncaseating granulomas. The granulomas appear as whorls of epithelioid cells surrounding multinucleated Langhans giant cells.
Definitive diagnosis requires tissue biopsy showing noncaseating granulomas. The granulomas appear as whorls of epithelioid cells surrounding multinucleated Langhans giant cells.
 Systemic evaluation and imaging studies: chest radiograph and/or high- resolution chest CT to identify hilar lymphadenopathy gallium scan (not recommended for routine screening); pulmonary function tests (reduction in diffusing capacity); bronchoalveolar lavage (elevated cd4/cd8 ratio); biopsy of the involved tissue; and/or brain MRI if neurosarcoidosis is suspected.
Systemic evaluation and imaging studies: chest radiograph and/or high- resolution chest CT to identify hilar lymphadenopathy gallium scan (not recommended for routine screening); pulmonary function tests (reduction in diffusing capacity); bronchoalveolar lavage (elevated cd4/cd8 ratio); biopsy of the involved tissue; and/or brain MRI if neurosarcoidosis is suspected.
Table 7-1. Diagnostic Criteria for Ocular Sarcoidosis from the International Workshop on Ocular Sarcoidosis, Tokyo, 2006
| Definite ocular sarcoidosis | Biopsy-supported diagnosis with compatible uveitis |
| Presumed ocular sarcoidosis | Biopsy not done; bilateral hilar lymphadenopathy with compatible uveitis |
| Probable ocular sarcoidosis | Biopsy not done; chest radiograph normal; 3 suggestive ocular signs and 2 positive investigational tests |
| Possible ocular sarcoidosis | Biopsy negative; 4 suggestive ocular signs and 2 positive investigations |
 Laboratory testing: Angiotensin-converting enzyme (ACE) is elevated in 75% of patients, lysozyme, hypercalcemia, hypercalciuria, anemia, elevated erythrocyte sedimentation rate (ESR)/C-reactive protein (CRP), anergy, elevated alkaline phosphatase.
Laboratory testing: Angiotensin-converting enzyme (ACE) is elevated in 75% of patients, lysozyme, hypercalcemia, hypercalciuria, anemia, elevated erythrocyte sedimentation rate (ESR)/C-reactive protein (CRP), anergy, elevated alkaline phosphatase.
 Fluorescein angiography can reveal retinal vascular leakage, early blockage and late staining of choroidal granulomas, retinal pigment epithelial (RPE) window defects, and cystoid macular edema (CME).
Fluorescein angiography can reveal retinal vascular leakage, early blockage and late staining of choroidal granulomas, retinal pigment epithelial (RPE) window defects, and cystoid macular edema (CME).
 None of the above is specific or diagnostic by itself; therefore, it is a clinical diagnosis, which is supported by imaging and laboratory testing. Tissue biopsy is required for definitive diagnosis.
None of the above is specific or diagnostic by itself; therefore, it is a clinical diagnosis, which is supported by imaging and laboratory testing. Tissue biopsy is required for definitive diagnosis.
Treatment
 Corticosteroids are the mainstay of therapy for both ocular and systemic sarcoidosis, and intravenous pulse steroids may be needed in some very severe cases.
Corticosteroids are the mainstay of therapy for both ocular and systemic sarcoidosis, and intravenous pulse steroids may be needed in some very severe cases.
 Sarcoidosis is often responsive to steroids; however, chronic uveitis often requires immunomodulatory therapy. Methotrexate, cyclosporine, mycophenolate mofetil, and infliximab have all been used successfully in ocular sarcoidosis.
Sarcoidosis is often responsive to steroids; however, chronic uveitis often requires immunomodulatory therapy. Methotrexate, cyclosporine, mycophenolate mofetil, and infliximab have all been used successfully in ocular sarcoidosis.
 Local treatment with topical drops, periocular steroids, and intraocular steroid implants can also be considered in select cases.
Local treatment with topical drops, periocular steroids, and intraocular steroid implants can also be considered in select cases.
Prognosis
 The prognosis is good if treated early.
The prognosis is good if treated early.
 African Americans tend to have more acute and severe disease, whereas Caucasians tend to have chronic, asymptomatic disease.
African Americans tend to have more acute and severe disease, whereas Caucasians tend to have chronic, asymptomatic disease.
 Chronic posterior or panuveitis, glaucoma, CME, older age at presentation, and delay in presentation to a sarcoid/uveitis subspecialist may confer a poor visual prognosis.
Chronic posterior or panuveitis, glaucoma, CME, older age at presentation, and delay in presentation to a sarcoid/uveitis subspecialist may confer a poor visual prognosis.
 Ocular complications can include glaucoma, cataract, CME, optic disk edema, occlusive vasculopathy, retinal or optic disk neovascularization, vitreous hemorrhage, and retinal detachment.
Ocular complications can include glaucoma, cataract, CME, optic disk edema, occlusive vasculopathy, retinal or optic disk neovascularization, vitreous hemorrhage, and retinal detachment.
REFERENCES
Baughman RP, Teirstein AS, Judson MA, et al. Case Control Etiologic Study of Sarcoidosis (ACCESS) research group. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001;164(10 Pt 1):1885–1889.
Dana MR, Merayo-Lloves J, Schaumberg DA, et al. Prognosticators for visual outcome in sarcoid uveitis. Ophthalmology. 1996;103(11):1846–1853.
Herbort CP, Rao NA, Mochizuki M. International criteria for the diagnosis of ocular sarcoidosis: results of the first International Workshop on Ocular Sarcoidosis (IWOS). Ocul Immunol Inflamm. 2009;17(3): 160–169.
Lobo A, Barton K, Minassian D, et al. Visual loss in sarcoid-related uveitis. Clin Experiment Ophthalmol. 2003;31(4):310–316.
Nussenblatt RB and Whitcup SM. Uveitis: Fundamentals and Clinical Practice. Philadelphia: Mosby; 2010: 278–288.
Figure 7-1. A. This patient with severe sarcoidosis has posterior uveitis with granulomas and intense exudates surrounding the vessels. Note the asymmetric involvement between the right and left eyes. The fluorescein angiogram of the left eye (lower right) demonstrates leakage and staining in the late frames. B. Exudates surrounding the vessels, also known as “candle-wax drippings” or “taches de bougie,” are seen inferiorly.
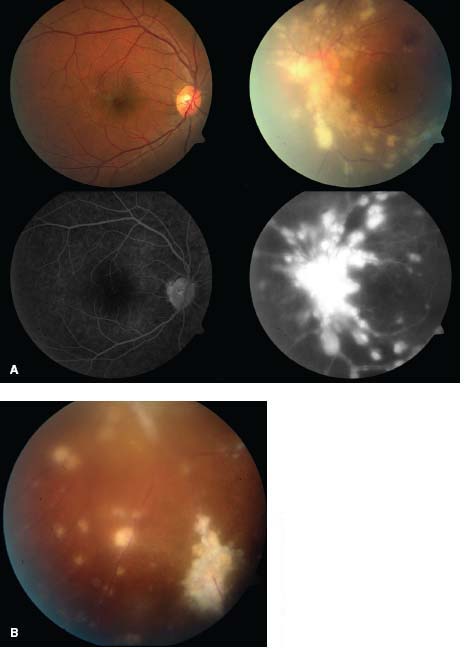
Figure 7-2. A. There are large, granulomatous-appearing (mutton-fat) keratic precipitates in this patient with sarcoidosis. B. Busacca nodules in the iris stroma are typically associated with diseases causing granulomatous uveitis such as sarcoidosis.
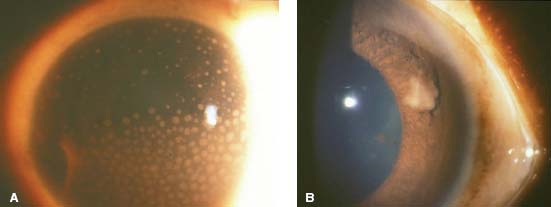
Figure 7-3. “Candle-wax drippings” along the major arcade vessels. (Courtesy of Sunir Garg, MD.)
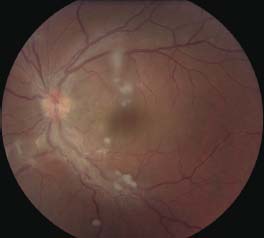
Figure 7-4. Sarcoidosis patient with intermediate uveitis with snowballs and retinal vascular sheathing inferiorly (magnified), as well as retinal vasculitis with leakage on fluorescein angiogram. Note that the extent of retinal vascular leakage is significantly more impressive than expected based on the fundus exam.
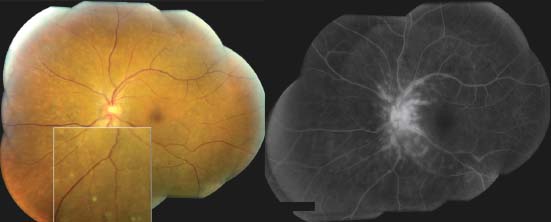
Figure 7-5. There are multiple skin nodules due to sarcoidosis in an African American patient with liver and ocular sarcoidosis. The inferior periorbital skin, the nose, elbow, and perioral skin are involved.
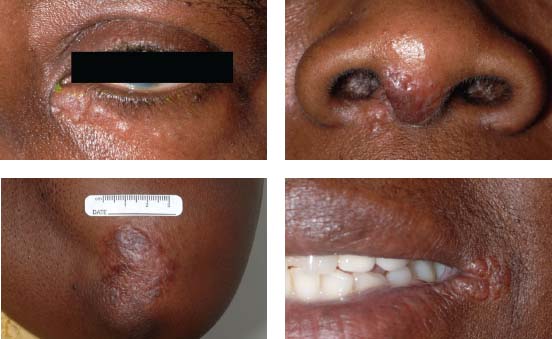
Figure 7-6. There are the typical punched-out chorioretinal lesions in a 65-year-old Caucasian patient with ocular sarcoidosis. These lesions are believed to be representative of atrophic Dalen-Fuchs nodules.
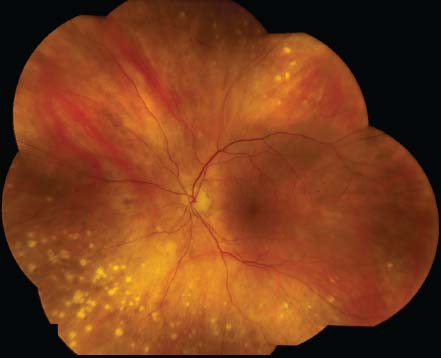
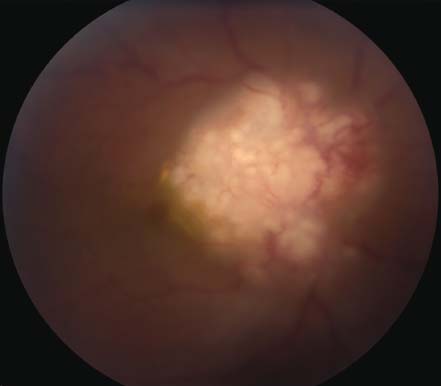
Figure 7-7. Optic nerve granuloma in the right eye of a 42-year-old African American patient with sarcoidosis.
SYMPATHETIC OPHTHALMIA
Rishi R. Doshi, S. R. Rathinam, and Emmett T. Cunningham, Jr.
Sympathetic ophthalmia is a bilateral granulomatous panuveitis that occurs when ocular surgery or ocular trauma to one eye (the exciting eye) incites inflammation both in the traumatized eye as well as in the fellow (sympathizing) eye.
Etiology and Epidemiology
 It has an estimated incidence of 0.03 in 100,000; the incidence is higher after vitreoretinal surgery (1 in 1152 procedures) compared to other ocular surgeries and surgery has surpassed trauma as the most common inciting event.
It has an estimated incidence of 0.03 in 100,000; the incidence is higher after vitreoretinal surgery (1 in 1152 procedures) compared to other ocular surgeries and surgery has surpassed trauma as the most common inciting event.
 Previously sequestered ocular antigens from the uvea, retina, or choroidal melanocytes stimulate an autoimmune response.
Previously sequestered ocular antigens from the uvea, retina, or choroidal melanocytes stimulate an autoimmune response.
 Historically, men and children were more commonly affected due to higher rates of trauma, but currently it occurs equally in both men and women with an increased incidence in elderly patients due to higher rates of surgery.
Historically, men and children were more commonly affected due to higher rates of trauma, but currently it occurs equally in both men and women with an increased incidence in elderly patients due to higher rates of surgery.
 It may occur after endophthalmitis, with an incidence from 1% to 11%.
It may occur after endophthalmitis, with an incidence from 1% to 11%.
 80% of cases occur within 3 months of surgery/trauma and 90% occur within 1 year, but it has been reported to occur anytime between 1 week to 66 years after injury.
80% of cases occur within 3 months of surgery/trauma and 90% occur within 1 year, but it has been reported to occur anytime between 1 week to 66 years after injury.
 There is an increased prevalence in patients with HLA-DR4 and HLA-A11.
There is an increased prevalence in patients with HLA-DR4 and HLA-A11.
Symptoms
 Patients often have a prodrome of tearing, photophobia, and blurry vision (loss of accommodation), which may progress to severe vision loss, pain, floaters, and photopsias in both eyes.
Patients often have a prodrome of tearing, photophobia, and blurry vision (loss of accommodation), which may progress to severe vision loss, pain, floaters, and photopsias in both eyes.
Signs (Figs. 7-8 to 7-12)
 Both eyes have inflammation with:
Both eyes have inflammation with:
 Anterior uveitis (55%); granulomatous KPs, iris thickening and synechiae, low or high intraocular pressure (from ciliary body shutdown or angle closure), vitritis (47%); disk edema (20%); serous retinal detachment (12%); choroiditis (8%); and macular edema (5%)
Anterior uveitis (55%); granulomatous KPs, iris thickening and synechiae, low or high intraocular pressure (from ciliary body shutdown or angle closure), vitritis (47%); disk edema (20%); serous retinal detachment (12%); choroiditis (8%); and macular edema (5%)
 Dalen-Fuchs nodules are yellow-white midequatorial RPE lesions composed of epithelioid cells and histiocytes (30% to 70%).
Dalen-Fuchs nodules are yellow-white midequatorial RPE lesions composed of epithelioid cells and histiocytes (30% to 70%).
 Late findings may include sunset glow fundus, optic atrophy, choroidal neovascularization, and phthisis.
Late findings may include sunset glow fundus, optic atrophy, choroidal neovascularization, and phthisis.
 Systemic findings may include cerebrospinal fluid pleocytosis, hearing disturbances, alopecia, poliosis, and vitiligo (but these are much more typical of VKH).
Systemic findings may include cerebrospinal fluid pleocytosis, hearing disturbances, alopecia, poliosis, and vitiligo (but these are much more typical of VKH).
Differential Diagnosis
 VKH (no history of penetrating trauma or surgery)
VKH (no history of penetrating trauma or surgery)
 Tuberculosis
Tuberculosis
 Sarcoidosis
Sarcoidosis
 Syphilis
Syphilis
 Intraocular lymphoma
Intraocular lymphoma
Diagnostic Evaluation
 On fluorescein angiography, patients have multiple pinpoint areas of hyperfluorescence with late leakage, choroidal lesions that block early and stain late, and late disk staining.
On fluorescein angiography, patients have multiple pinpoint areas of hyperfluorescence with late leakage, choroidal lesions that block early and stain late, and late disk staining.
 Optical coherence tomography can show multifocal serous retinal detachments.
Optical coherence tomography can show multifocal serous retinal detachments.
 B-scan ultrasonography demonstrates choroidal thickening and serous retinal detachments.
B-scan ultrasonography demonstrates choroidal thickening and serous retinal detachments.
 Histopathology characteristically demonstrates a diffuse granulomatous nonnecrotizing panuveitis with thickening of choroid and early sparing of choriocapillaris.
Histopathology characteristically demonstrates a diffuse granulomatous nonnecrotizing panuveitis with thickening of choroid and early sparing of choriocapillaris.
Treatment
 First-line therapy is high-dose oral corticosteroids (1 to 2 mg/kg/day), with a slow taper.
First-line therapy is high-dose oral corticosteroids (1 to 2 mg/kg/day), with a slow taper.
 Topical corticosteroids and cycloplegic/mydriatic agents can help alleviate some of the more acute symptoms.
Topical corticosteroids and cycloplegic/mydriatic agents can help alleviate some of the more acute symptoms.
 Corticosteroid-sparing immunosuppressive agents are typically required, as most patients need long-term treatment.
Corticosteroid-sparing immunosuppressive agents are typically required, as most patients need long-term treatment.
 A fluocinolone acetonide implant (Retisert) may be useful in select cases.
A fluocinolone acetonide implant (Retisert) may be useful in select cases.
 Enucleation of the exciting eye within 2 weeks of the inciting event may decrease the risk of sympathetic ophthalmia; once sympathetic ophthalmia occurs, enucleation should be considered only in a blind, painful eye.
Enucleation of the exciting eye within 2 weeks of the inciting event may decrease the risk of sympathetic ophthalmia; once sympathetic ophthalmia occurs, enucleation should be considered only in a blind, painful eye.
Prognosis
 It is a vision-threatening disease, with only slightly more than half of treated patients retaining visual acuity ≥20/40. One-fourth of patients have vision worse than 20/200.
It is a vision-threatening disease, with only slightly more than half of treated patients retaining visual acuity ≥20/40. One-fourth of patients have vision worse than 20/200.
 Complications include cataract and glaucoma.
Complications include cataract and glaucoma.
 Predictors of a poor outcome include a traumatic etiology, active, uncontrolled intraocular inflammation, and an exudative retinal detachment.
Predictors of a poor outcome include a traumatic etiology, active, uncontrolled intraocular inflammation, and an exudative retinal detachment.
 Long-term visual loss may result from chorioretinal scars, chronic macular edema, and choroidal neovascularization
Long-term visual loss may result from chorioretinal scars, chronic macular edema, and choroidal neovascularization
REFERENCES
Castiblanco CP, Adelman RA. Sympathetic ophthalmia. Graefes Arch Clin Exp Ophthalmol. 2009;247:289–302.
Galor A, Davis JL, Flynn HW, et al. Sympathetic ophthalmia: Incidence of ocular complications and vision loss in the sympathizing eye. Am J Ophthalmol. 2009;148:704–710.
Kilmartin DJ, Dick AD, Forrester JV. Prospective surveillance of sympathetic ophthalmia in the United Kingdom and Republic of Ireland. Br J Ophthalmol. 2000;84:259–263.
Rathinam SR, Rao NA. Sympathetic ophthalmia following postoperative bacterial endophthalmitis: a clinicopathologic study. Am J Ophthalmol. 2006;141(3):498–507.
Figure 7-8. Phthisis bulbi following a ruptured globe in a patient with sympathetic ophthalmia.
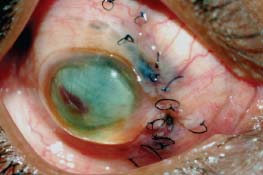
Figure 7-9. B-scan ultrasound showing a chronic retinal detachment and choroidal thickening in a patient with sympathetic ophthalmia.
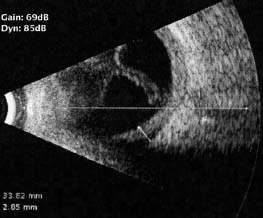
Figure 7-10. Characteristic posterior segment findings in two patients who developed sympathetic ophthalmia weeks after uncomplicated pars plana vitrectomy. Color fundus photograph (A) and midphase fluorescein angiogram (B) from the same patient 2 months following vitrectomy shows a macular serous detachment with multiple, pin-point areas of leakage through the retinal pigment epithelium. (C) Color fundus photographs taken approximately 6 months following vitrectomy shows moderate vitreous inflammation and active chorioretinal infiltrates. (D) Photograph of the same patient taken approximately 18 months following vitrectomy, after the patient’s inflammation had been controlled with high dose systemic corticosteroids followed by noncorticosteroid immunosuppressive agents. Although histopathologically unverified, many would describe these infiltrates clinically as Dalen-Fuchs nodules. (Reproduced from Doshi RR, Arevalo JF, Flynn HW Jr, et al. Evaluating exaggerated, prolonged, or delayed postoperative intraocular inflammation. Am J Ophthalmol. 2010, with permission of Elsevier.)
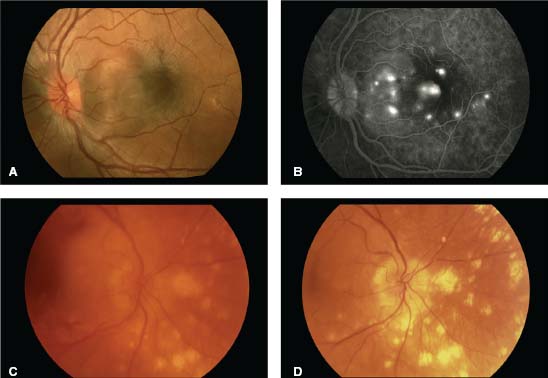
Figure 7-11. Bullous serous retinal detachment in a patient with sympathetic ophthalmia.
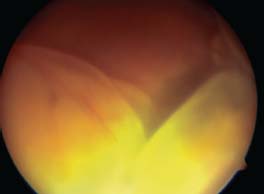
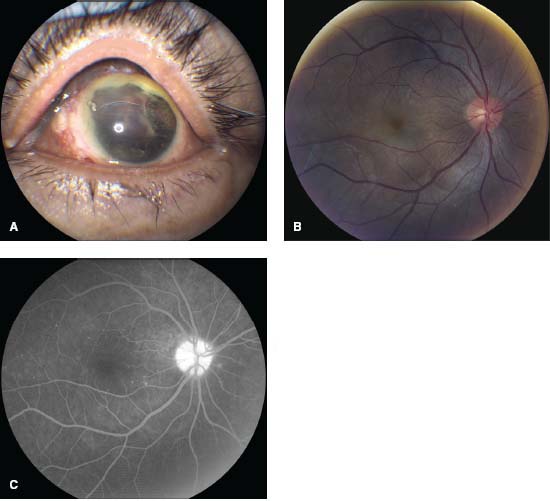
Figure 7-12. A. This patient underwent ruptured globe repair following blunt trauma. B. Several months later, the patient returned complaining of photophobia and blurred vision in the previously healthy right eye. There is disk hyperemia, mild macular striations due to subretinal fluid, and deep, choroidal yellow plaques characteristic of sympathetic ophthalmia. C. The fluorescein angiogram demonstrates disk hyperfluorescence and pinpoint leakage and staining from the areas of choroiditis. (Courtesy Allen Chiang, MD, and Andre Witkin, MD.)
VOGT-KOYANAGI-HARADA SYNDROME
Nupura Krishnadev, Robert Nussenblatt, and H. Nida Sen
Vogt-Koyanagi-Harada (VHK) syndrome, or uveomeningitis, is a bilateral granulomatous panuveitis with skin, meningeal, and auditory-vestibular involvement.
Epidemiology and Etiology
 More commonly seen in patients from Japan and Latin America. It also occurs in African Americans and in persons with Native American ancestry.
More commonly seen in patients from Japan and Latin America. It also occurs in African Americans and in persons with Native American ancestry.
 It is most common in the second to fourth decade of life.
It is most common in the second to fourth decade of life.
 There is a slight female preponderance.
There is a slight female preponderance.
 VKH has a genetic predisposition, and has been associated with HLA-DR4, HLA-DRw53, and HLA-DRB1*0405.
VKH has a genetic predisposition, and has been associated with HLA-DR4, HLA-DRw53, and HLA-DRB1*0405.
 It tends to occur in the spring and fall.
It tends to occur in the spring and fall.
 Unlike sympathetic ophthalmia, patients with VKH have no history of penetrating ocular trauma or surgery.
Unlike sympathetic ophthalmia, patients with VKH have no history of penetrating ocular trauma or surgery.
Symptoms
 Blurry vision, floaters
Blurry vision, floaters
 Headache, vertigo, neck stiffness
Headache, vertigo, neck stiffness
 Dysacusis and tinnitus
Dysacusis and tinnitus
 Skin changes: vitiligo, (skin depigmentation), poliosis (hair whitening), and alopecia in the later stages (Fig. 7-13)
Skin changes: vitiligo, (skin depigmentation), poliosis (hair whitening), and alopecia in the later stages (Fig. 7-13)
Signs
 The American Uveitis Society (AUS) and the International Uveitis Society have established criteria for diagnosis. The AUS criteria for complete VKH are shown in Table 7-2.
The American Uveitis Society (AUS) and the International Uveitis Society have established criteria for diagnosis. The AUS criteria for complete VKH are shown in Table 7-2.
Differential Diagnosis
 Sympathetic ophthalmia (these patients have a history of penetrating trauma)
Sympathetic ophthalmia (these patients have a history of penetrating trauma)
 Bullous central serous chorioretinopathy (these patients have no inflammation)
Bullous central serous chorioretinopathy (these patients have no inflammation)
 Posterior scleritis (eyes have scleral thickening on B-scan ultrasonography)
Posterior scleritis (eyes have scleral thickening on B-scan ultrasonography)
 Sarcoidosis
Sarcoidosis
 Syphilis
Syphilis
 Lyme disease
Lyme disease
 Ocular lymphoma
Ocular lymphoma
 Uveal metastasis
Uveal metastasis
Diagnostic Evaluation (Figs. 7-14 to 7-18)
 Fluorescein angiography: Early in the angiogram, there are multiple pinpoint areas of hyperfluorescence at the level of the RPE, which pool in the later frames. Late staining of the disk is often present.
Fluorescein angiography: Early in the angiogram, there are multiple pinpoint areas of hyperfluorescence at the level of the RPE, which pool in the later frames. Late staining of the disk is often present.
 B-scan ultrasonography: Demonstrates nonspecific choroidal thickening and serous retinal detachments but can be helpful to rule out posterior scleritis and neoplastic disorders.
B-scan ultrasonography: Demonstrates nonspecific choroidal thickening and serous retinal detachments but can be helpful to rule out posterior scleritis and neoplastic disorders.
 Lumbar puncture: The majority of patients will have pleocytosis (but usually this is not needed).
Lumbar puncture: The majority of patients will have pleocytosis (but usually this is not needed).
 Laboratory tests to rule out other diseases such as sarcoid and syphilis as needed.
Laboratory tests to rule out other diseases such as sarcoid and syphilis as needed.
Treatment
 These patients need prompt and aggressive systemic therapy. Corticosteroids are the first-line agent, with some patients requiring 100 mg of prednison daily, with a slow taper.
These patients need prompt and aggressive systemic therapy. Corticosteroids are the first-line agent, with some patients requiring 100 mg of prednison daily, with a slow taper.
 Most patients will need immunosuppression for 1 year.
Most patients will need immunosuppression for 1 year.
 If higher doses of prednisone are used for more than 2 to 3 months, steroid-sparing agents such as cyclosporine, tacrolimus, azathioprine, and mycophenolate mofetil should be considered.
If higher doses of prednisone are used for more than 2 to 3 months, steroid-sparing agents such as cyclosporine, tacrolimus, azathioprine, and mycophenolate mofetil should be considered.
Table 7-2. Diagnostic Criteria for VKH
| 1. No history of penetrating ocular trauma |
| 2. No evidence of other ocular disease |
| 3. Bilateral ocular involvement |
a. Early |
i. Diffuse choroiditis presenting as |
1. Focal areas of subretinal fluid, or |
2. Bullous serous retinal detachments |
ii. If equivocal fundus findings, then must have both of below: |
1. IVFA showing delayed choroidal perfusion, pinpoint leakage, pooling within the subretinal space, and optic nerve staining |
2. Ultrasound with diffuse choroidal thickening without posterior scleritis |
b. Late |
i. History suggestive of above, or both ii and iii below, or multiple signs from iii: |
ii. Ocular depigmentation |
1. Sunset glow fundus, or |
2. Sugiura sign |
iii. Other ocular signs |
1. Nummular chorioretinal depigmented scars, or |
2. RPE clumping, or |
3. Recurrent or chronic anterior uveitis |
| 4. Neurologic/auditory findings |
a. Meningismus, or |
b. Tinnitus, or |
c. CSF pleocytosis |
| 5. Integumentary (skin) findings |
a. Alopecia, or |
b. Poliosis, or |
c. Vitiligo |
| Complete VKH requires criteria 1 to 5 |
| Incomplete VKH requires 1 to 3, and either 4 or 5 |
| Probable VKH requires 1 to 3 (isolated ocular disease) |
Prognosis
 The prognosis is generally good with prompt and aggressive therapy.
The prognosis is generally good with prompt and aggressive therapy.
 In addition to the development of cataracts and glaucoma, subretinal fibrosis, and choroidal neovascular membranes may occur.
In addition to the development of cataracts and glaucoma, subretinal fibrosis, and choroidal neovascular membranes may occur.
REFERENCES
Rao NA, Gupta A, Dustin L, et al. Frequency of distinguishing clinical features in Vogt-Koyanagi-Harada disease. Ophthalmology. 2010;117(3):591–599, 599.e1. Epub 2009 Dec 24.
Read RW, Holland GN, Rao NA, et al. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: Report of an International Committee on Nomenclature. Am J Ophthalmol. 2001;131:647–652.
Yamaguchi Y, Otani T, Kishi S. Tomographic features of serous retinal detachment with multilobular dye pooling in acute Vogt-Koyanagi-Harada disease. Am J Ophthalmol. 2007;144(2):260–265. Epub 2007 May 29.
Figure 7-13. A. Vitiligo. This African American patient with VKH had extensive areas of skin depigmentation. B. Poliosis. A single white eyelash (poliosis) was found in this African American patient with VKH.
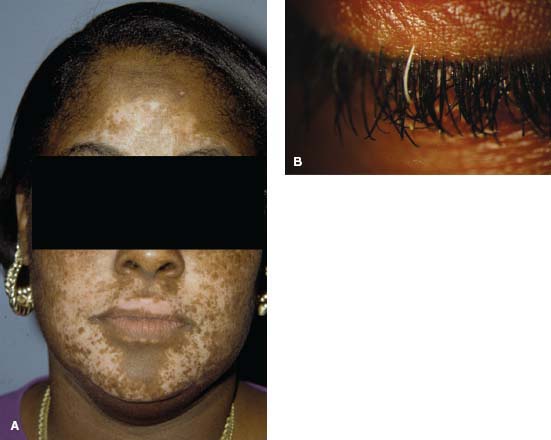
Figure 7-14. Serous retinal detachments in early VKH. A. Multiple small serous detachments in the right macula of an African American male presenting with a 5-day history of blurred vision. B. Fundus autofluorescence shows alterations in the RPE. C. Fluorescein angiogram (FA) in the early arteriovenous phase reveals the classic finding of multiple areas of pinpoint leakage. D. The late phase of the FA demonstrates pooling of fluorescein in the areas of serous retinal detachments.
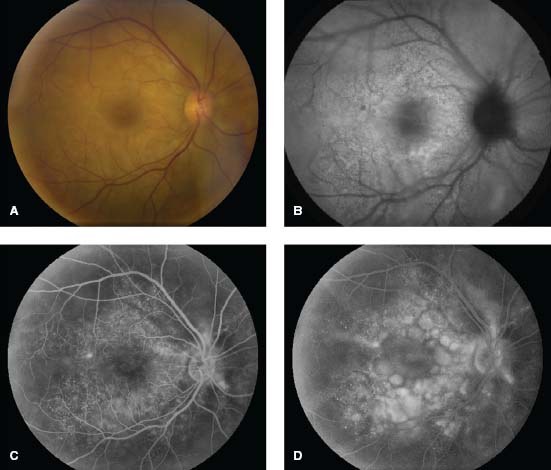
Figure 7-15. A. This 43-year-old woman from Central America has multiple serous retinal detachments and deep yellow lesions consistent with choroiditis. B. The FA shows multiple pinpoint areas of leakage with pooling of dye in the areas of serous retinal detachment. There is also staining of the optic disk. (Courtesy Sunir Garg, MD, and MidAtlantic Retina, the Retina Service of Wills Eye Institute.)
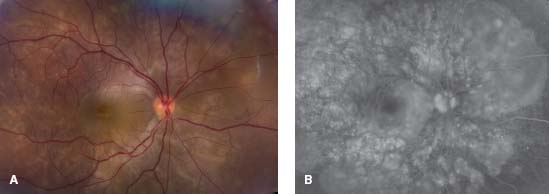
Figure 7-16. Optical coherence tomography (OCT) demonstrates a serous retinal detachment withsubretinal septae, a finding suggestive of VKH. (Courtesy MidAtlantic Retina, The Retina Service of Wills Eye Institute.)
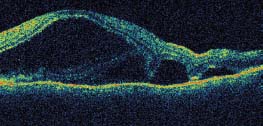
Figure 7-17. Sunset glow fundus in late VKH. The orange-red color of the fundus, due to loss of choroidal melanocytes in this patient with Native American ancestry, can develop 2 to 6 months after disease onset. Changes in the RPE are also visible.
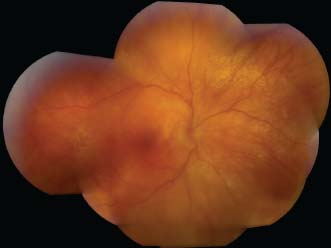
Figure 7-18. Subretinal fibrosis in late VKH. Note the extensive fibrosis and pigmentary changes in an advanced case of VKH with a history of poorly controlled uveitis.
OCULAR COMPLICATIONS OF RHEUMATOID ARTHRITIS
Bhupesh Bagga and Virender S. Sangwan
Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disease that predominantly affects the peripheral joints. Patients with rheumatoid arthritis can also have extra-articular systemic and ocular in-volvement.
Epidemiology and Etiology
 RA is distributed worldwide, with a prevalence between 0.3% and 1.5%.
RA is distributed worldwide, with a prevalence between 0.3% and 1.5%.
 Women develop RA more often than men by a 3:1 ratio.
Women develop RA more often than men by a 3:1 ratio.
 Although the disease can occur at any point, the incidence increases with advancing age.
Although the disease can occur at any point, the incidence increases with advancing age.
 Activated T cells and antigen-antibody complexes cause a microvasculitis that affects joint synovial tissues as well as extra-articular tissues. Immune complex deposition and activation of complement plays a pivotal role in the systemic and ocular pathophysiology.
Activated T cells and antigen-antibody complexes cause a microvasculitis that affects joint synovial tissues as well as extra-articular tissues. Immune complex deposition and activation of complement plays a pivotal role in the systemic and ocular pathophysiology.
Symptoms
 Patients may complain of a foreign body sensation, and photophobia. A red eye due to episcleritis and dry eye are both common in RA.
Patients may complain of a foreign body sensation, and photophobia. A red eye due to episcleritis and dry eye are both common in RA.
 Patients with scleritis complain of a deep, boring, periocular pain that radiates to the brow and temple.
Patients with scleritis complain of a deep, boring, periocular pain that radiates to the brow and temple.
Signs
 Ocular (Figs. 7-19 to 7-23)
Ocular (Figs. 7-19 to 7-23)
 Keratoconjunctivitis sicca (dry eye): Rapid tear film breakup time and devitalized corneal epithelium on staining are seen. Schirmer testing demonstrates decreased tear production.
Keratoconjunctivitis sicca (dry eye): Rapid tear film breakup time and devitalized corneal epithelium on staining are seen. Schirmer testing demonstrates decreased tear production.
 Sterile corneal infiltrates, and corneal edema, opacification, vascularization, and/or thinning with marginal furrowing can occur.
Sterile corneal infiltrates, and corneal edema, opacification, vascularization, and/or thinning with marginal furrowing can occur.
 Peripheral ulcerative keratitis is a more aggressive form of perilimbal vasculitis.
Peripheral ulcerative keratitis is a more aggressive form of perilimbal vasculitis.
 Episcleritis is characterized by dilated episcleral vessels.
Episcleritis is characterized by dilated episcleral vessels.
 Patients with scleritis have a dilated superficial and deep episcleral vascular plexus that imparts a violaceous hue to the sclera. Unlike in episcleritis, the dilated vessels lose their normal radial architecture.
Patients with scleritis have a dilated superficial and deep episcleral vascular plexus that imparts a violaceous hue to the sclera. Unlike in episcleritis, the dilated vessels lose their normal radial architecture.
 Diffuse anterior scleritis is a more common form and has a better prognosis than other types of scleritis including nodular scleritis, anterior necrotizing scleritis, necrotizing scleritis without inflammation (scleromalacia perforans), and posterior scleritis.
Diffuse anterior scleritis is a more common form and has a better prognosis than other types of scleritis including nodular scleritis, anterior necrotizing scleritis, necrotizing scleritis without inflammation (scleromalacia perforans), and posterior scleritis.
 Systemic
Systemic
 Patients initially develop fatigue, morning stiffness, and myalgias. The typical finding is polyarthritis that primarily affects the hands, feet, and cervical vertebrae.
Patients initially develop fatigue, morning stiffness, and myalgias. The typical finding is polyarthritis that primarily affects the hands, feet, and cervical vertebrae.
 Patients may also develop anemia, pericarditis, pleuritis, glomerulonephritis, neuropathy, and vasculitis of the skin (Fig. 7-24).
Patients may also develop anemia, pericarditis, pleuritis, glomerulonephritis, neuropathy, and vasculitis of the skin (Fig. 7-24).
Differential Diagnosis
 Sjögren’s syndrome
Sjögren’s syndrome
 Reiter’s syndrome
Reiter’s syndrome
 Enteropathic arthritis, including Crohn’s disease and reactive arthritis
Enteropathic arthritis, including Crohn’s disease and reactive arthritis
 Psoriatic arthritis
Psoriatic arthritis
 Systemic lupus erythematosus
Systemic lupus erythematosus
 Sarcoidosis
Sarcoidosis
 Wegener’s granulomatosis
Wegener’s granulomatosis
 Dermatomyositis
Dermatomyositis
 Polyarteritis nodosa
Polyarteritis nodosa
Diagnostic Evaluation
 Specific markers: Rheumatoid factor (RF), antinuclear antibodies (ANA), antineutrophil cytoplasmic antibody (ANCA), anti-SS-A, anti-SS-B antibodies, and antibodies to cyclic citrullinated peptides
Specific markers: Rheumatoid factor (RF), antinuclear antibodies (ANA), antineutrophil cytoplasmic antibody (ANCA), anti-SS-A, anti-SS-B antibodies, and antibodies to cyclic citrullinated peptides
 Nonspecific markers: Complete blood count, chest radiograph, ESR, CRP, liver function tests, kidney function tests
Nonspecific markers: Complete blood count, chest radiograph, ESR, CRP, liver function tests, kidney function tests
Treatment
 Systemic disease: Three major classes of drugs are used: NSAIDs, disease-modifying antirheumatic drugs (DMARDs), and corticosteroids.
Systemic disease: Three major classes of drugs are used: NSAIDs, disease-modifying antirheumatic drugs (DMARDs), and corticosteroids.
 Ocular disease
Ocular disease
 For cases of dry eye, artificial tear substitutes and lubricating ointments, topical steroids, or topical cyclosporin A 2% drops should be used. Temporary punctal plugs and/or permanent punctal occlusion with cautery or permanent tarsorrhaphy are also useful modalities for maintaining adequate ocular lubrication.
For cases of dry eye, artificial tear substitutes and lubricating ointments, topical steroids, or topical cyclosporin A 2% drops should be used. Temporary punctal plugs and/or permanent punctal occlusion with cautery or permanent tarsorrhaphy are also useful modalities for maintaining adequate ocular lubrication.
 Keratitis: Topical and oral corticosteroids and topical 2% cyclosporin A drops may play a role in the treatment of RA-associated keratitis, particularly nonulcerative keratitis and infiltrative PUK. However, more severe cases may require systemic immunosuppression.
Keratitis: Topical and oral corticosteroids and topical 2% cyclosporin A drops may play a role in the treatment of RA-associated keratitis, particularly nonulcerative keratitis and infiltrative PUK. However, more severe cases may require systemic immunosuppression.
 Scleritis: In mild cases, NSAIDS can be used along with, or instead of, low-dose oral steroids. Moderate to severe disease, including necrotizing scleritis and PUK, require systemic treatment. If patients cannot achieve disease remission with less than 10 mg of prednisone daily, steroid-sparing agents should be used. Antimetabolites (methotrexate, azathioprine, and mycophenolate mofetil), calcineurin inhibitors (cyclosporin, and tacrolimus), and biologic agents (infliximab, adalimumab, and rituximab) have all been used with varying degrees of success. Although etanercept has good efficacy for the treatment of systemic RA, it is less efficacious for the treatment of the ocular disease than are the other biologic agents.
Scleritis: In mild cases, NSAIDS can be used along with, or instead of, low-dose oral steroids. Moderate to severe disease, including necrotizing scleritis and PUK, require systemic treatment. If patients cannot achieve disease remission with less than 10 mg of prednisone daily, steroid-sparing agents should be used. Antimetabolites (methotrexate, azathioprine, and mycophenolate mofetil), calcineurin inhibitors (cyclosporin, and tacrolimus), and biologic agents (infliximab, adalimumab, and rituximab) have all been used with varying degrees of success. Although etanercept has good efficacy for the treatment of systemic RA, it is less efficacious for the treatment of the ocular disease than are the other biologic agents.
 Nodular scleritis may be treated with injection of triamcinolone over the lesion, and some authors have reported success in treating certain types of anterior scleritis with periocular steroid injections.
Nodular scleritis may be treated with injection of triamcinolone over the lesion, and some authors have reported success in treating certain types of anterior scleritis with periocular steroid injections.
Prognosis
If treated early, long-term remission can often be obtained. Acute exacerbations of the disease can be avoided with regular monitoring and titration of the dose of the immunosuppressive agent.
REFERENCES
Albini TA, Zamir E, Read RW, et al. Evaluation of subconjunctival triamcinolone for nonnecrotizing anterior scleritis. Ophthalmology. 2005;112(10):1814–1820.
Chauhan S, Kamal A, Thompson RN, et al. Rituximab for treatment of scleritis associated with rheumatoid arthritis. Br J Ophthalmol. 2009;93(7):984–985.
Gangaputra S, Newcomb CW, Liesegang TL, et al. Systemic Immunosuppressive Therapy for Eye Diseases Cohort Study. Methotrexate for ocular inflammatory diseases. Ophthalmology. 2009;116(11):2188–2198.
Figure 7-19. Diffuse scleritis is characterized by deep, tortuous, dilated scleral vessels. There is also peripheral corneal melting with iris prolapse in this case. (Courtesy of S. R. Rathinam.)
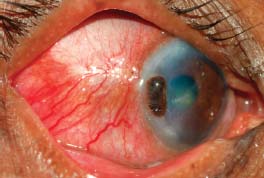
Figure 7-20. There is peripheral ulcerative keratitis in a patient with active, previously undiagnosed rheumatoid arthritis. (Courtesy of S. R. Rathinam.)
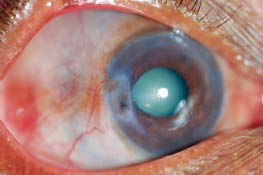
Figure 7-21. There are several large, injected nodules as well as diffuse scleritis in this patient with nodular scleritis. Posterior synechiae and a cataract are present. (Courtesy of S. R. Rathinam.)
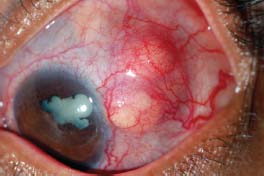
Figure 7-22. A. Significant scleral thinning with uveal prolapse. The lack of injection is typical of scleromalacia perforans. (Courtesy of Sunir J. Garg.) B. Significant scleral thinning with an inferior scleral melt. Compared with patients with necrotizing scleritis, patients with scleromalacia perforans have relatively little scleral redness and often have minimal to no discomfort. (Courtesy of S. R. Rathinam.)
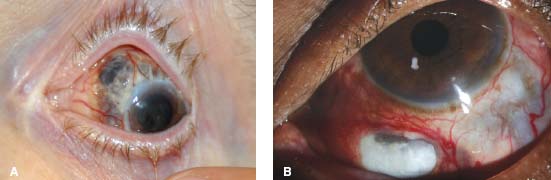
Figure 7-23. A, B. Rheumatoid arthritis is a polyarthritis which affects the small joints of the hands and feet more frequently than larger joints. The synovial inflammation leads to joint destruction. These patients have the typical finger joint deformities seen in patients with rheumatoid arthritis. Prompt and aggressive therapy can dramatically reduce this debilitating aspect of RA. C. Similar joint destruction can also occur in the feet. (Courtesy of S. R. Rathinam.)
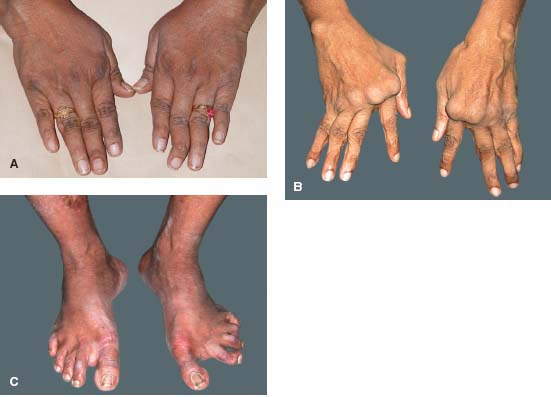
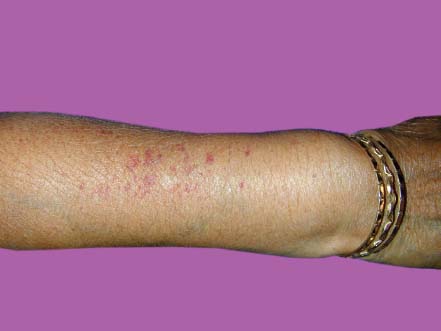
Figure 7-24. This patient has vasculitis of the skin, which can be seen in some patients with rheumatoid arthritis. (Courtesy of S. R. Rathinam.)
BEHÇET’S DISEASE
H. Nida Sen
Behçet’s disease (BD), or Adamantiades-Behçet’s disease, is a chronic, relapsing multisystem inflammatory disorder of unknown etiology that is characterized by intraocular inflammation, oral and mucosal ulcerations, and skin lesions.
Epidemiology and Etiology
 It is more commonly seen in patients from the Mediterranean basin and Japan, along the route of the ancient “silk road.” It also occurs in persons with Native American ancestry.
It is more commonly seen in patients from the Mediterranean basin and Japan, along the route of the ancient “silk road.” It also occurs in persons with Native American ancestry.
 In the United States, the prevalence is 8.6 in 100,000 persons, and is as high as 25 to 400 in 100,000 in Mediterranean countries.
In the United States, the prevalence is 8.6 in 100,000 persons, and is as high as 25 to 400 in 100,000 in Mediterranean countries.
 Ocular involvement occurs in approximately 70% of patients with Behçet’s disease and is typically bilateral (>70%). Posterior/panuveitis is the most common form of uveitis in these patients.
Ocular involvement occurs in approximately 70% of patients with Behçet’s disease and is typically bilateral (>70%). Posterior/panuveitis is the most common form of uveitis in these patients.
 Ocular disease can be the presenting symptom in 10% to 35% of patients and is characterized by recurrent, explosive exacerbations of intraocular inflammation.
Ocular disease can be the presenting symptom in 10% to 35% of patients and is characterized by recurrent, explosive exacerbations of intraocular inflammation.
 It typically affects young adults (mean age at onset is 25 to 35 years), but it may also occur in children.
It typically affects young adults (mean age at onset is 25 to 35 years), but it may also occur in children.
 Men are slightly more affected than women.
Men are slightly more affected than women.
 Behçet’s is a multifactorial disease: Genetic factors, environmental factors, infectious agents, and immunologic mechanisms all have been suggested.
Behçet’s is a multifactorial disease: Genetic factors, environmental factors, infectious agents, and immunologic mechanisms all have been suggested.
 Affected patients are thought to have a genetic predisposition, and it has been associated with HLA-B51.
Affected patients are thought to have a genetic predisposition, and it has been associated with HLA-B51.
Symptoms
 Redness, blurry vision, floaters
Redness, blurry vision, floaters
 Oral ulcers, genital ulcers, skin changes
Oral ulcers, genital ulcers, skin changes
Signs
 The International Society for Behçet’s Disease and the Japanese Behçet’s Disease Research Committee developed criteria for the diagnosis (Table 7-3).
The International Society for Behçet’s Disease and the Japanese Behçet’s Disease Research Committee developed criteria for the diagnosis (Table 7-3).
 Systemic manifestations: oral ulcers are the most common manifestation occurring in 98% of patients, followed by skin lesions (90%), and genital ulcers (77% to 85%).
Systemic manifestations: oral ulcers are the most common manifestation occurring in 98% of patients, followed by skin lesions (90%), and genital ulcers (77% to 85%).
 Ocular signs usually manifest as an explosive anterior uveitis typically with a hypopyon. Vitritis, necrotizing retinitis, retinal vasculitis, retinal hemorrhages, retinal edema, vitreous hemorrhage, capillary dropout, retinal neovascularization, and vitreous hemorrhage also occur.
Ocular signs usually manifest as an explosive anterior uveitis typically with a hypopyon. Vitritis, necrotizing retinitis, retinal vasculitis, retinal hemorrhages, retinal edema, vitreous hemorrhage, capillary dropout, retinal neovascularization, and vitreous hemorrhage also occur.
 Rare ocular manifestations include episcleritis, filamentary keratitis, conjunctivitis and extraocular muscle paralysis secondary to neuro-Behçet’s disease.
Rare ocular manifestations include episcleritis, filamentary keratitis, conjunctivitis and extraocular muscle paralysis secondary to neuro-Behçet’s disease.
 Between episodes, the ocular exam may be unremarkable.
Between episodes, the ocular exam may be unremarkable.
Differential Diagnosis
 HLA-B27–associated anterior uveitis with hypopyon
HLA-B27–associated anterior uveitis with hypopyon
 Pseudohypopyon
Pseudohypopyon
 Viral retinitis (specifically CMV, VZV, HSV)
Viral retinitis (specifically CMV, VZV, HSV)
 Toxoplasmosis
Toxoplasmosis
 Ocular lymphoma
Ocular lymphoma
 Lupus retinal vasculitis
Lupus retinal vasculitis
 Polyarteritis nodosa (PAN)
Polyarteritis nodosa (PAN)
 Sarcoidosis
Sarcoidosis
 Wegener’s granulomatosis–associated vasculitis
Wegener’s granulomatosis–associated vasculitis
Diagnostic Evaluation (Figs. 7-25 to 7-33)
 Fluorescein angiography demonstrates early blockage in areas of retinitis with late staining, staining and leakage of retinal vessels in the mid to late phases, and areas of capillary drop-out.
Fluorescein angiography demonstrates early blockage in areas of retinitis with late staining, staining and leakage of retinal vessels in the mid to late phases, and areas of capillary drop-out.
 Indocyanine green (ICG) angiography demonstrates areas of hyper- and hypofluorescence.
Indocyanine green (ICG) angiography demonstrates areas of hyper- and hypofluorescence.
 Presence of HLA-B51 is supportive of the diagnosis, but not diagnostic.
Presence of HLA-B51 is supportive of the diagnosis, but not diagnostic.
 A positive pathergy test and elevated ESR, CRP, and white blood count (WBC) are also suggestive of the diagnosis, but not diagnostic.
A positive pathergy test and elevated ESR, CRP, and white blood count (WBC) are also suggestive of the diagnosis, but not diagnostic.
 Histopathology demonstrates leukocytoclastic and monocytic occlusive vasculitis.
Histopathology demonstrates leukocytoclastic and monocytic occlusive vasculitis.
Treatment
 Prompt and aggressive systemic therapy with corticosteroids as the first-line agent is critical. Initially, some patients may need IV pulse steroids to control acute inflammation.
Prompt and aggressive systemic therapy with corticosteroids as the first-line agent is critical. Initially, some patients may need IV pulse steroids to control acute inflammation.
 Most patients will need immunosuppressive agents, which typically include cyclosporine, azathioprine, mycophenolate mofetil, alkylating agents (i.e., Cytoxan), infliximab, interferon-alpha, or a combination of these agents.
Most patients will need immunosuppressive agents, which typically include cyclosporine, azathioprine, mycophenolate mofetil, alkylating agents (i.e., Cytoxan), infliximab, interferon-alpha, or a combination of these agents.
 The main goal of treatment is to decrease the frequency and severity of the inflammatory episodes.
The main goal of treatment is to decrease the frequency and severity of the inflammatory episodes.
Prognosis
 The visual prognosis depends upon the involvement of the posterior segment, timeliness of therapy, and sustained control of inflammation.
The visual prognosis depends upon the involvement of the posterior segment, timeliness of therapy, and sustained control of inflammation.
 Younger age at onset, poor vision at presentation, posterior segment disease, persistent inflammatory activity, posterior synechiae, elevated intraocular pressure, and hypotony are poor prognostic indicators.
Younger age at onset, poor vision at presentation, posterior segment disease, persistent inflammatory activity, posterior synechiae, elevated intraocular pressure, and hypotony are poor prognostic indicators.
 With prompt and aggressive therapy, structural complications and visual impairment can be minimized.
With prompt and aggressive therapy, structural complications and visual impairment can be minimized.
 Complications are frequent and occur in 60% to 90% of patients. In addition to the development of cataracts and glaucoma, posterior synechiae, macular edema, epiretinal membrane, retinal neovascularization, vitreous hemorrhage, retinal and/or optic atrophy, branch retinal vein occlusion, branch retinal artery occlusion, and retinal detachment can occur.
Complications are frequent and occur in 60% to 90% of patients. In addition to the development of cataracts and glaucoma, posterior synechiae, macular edema, epiretinal membrane, retinal neovascularization, vitreous hemorrhage, retinal and/or optic atrophy, branch retinal vein occlusion, branch retinal artery occlusion, and retinal detachment can occur.
 Significant vision loss (<20/200) occurs in 20% to 30% of patients within 5 years of disease onset.
Significant vision loss (<20/200) occurs in 20% to 30% of patients within 5 years of disease onset.
REFERENCES
International Study Group for Behçet’s Disease, Criteria for diagnosis of Behçet’s disease, Lancet 335 (1990): 1078–1080.
Kaçmaz RO, Kempen JH, Newcomb C, et al. Systemic Immunosuppressive Therapy for Eye Diseases Cohort Study Group. Ocular inflammation in Behçet disease: incidence of ocular complications and of loss of visual acuity. Am J Ophthalmol. 2008;146(6):828–836.
Nussenblatt RB, Whitcup SM. Uveitis: Fundamentals and Clinical Practice. 4th ed. Philadelphia: Mosby Elsevier; 2010.
Tugal-Tutkun I, Onal S, Altan-Yaycioglu R, et al. Uveitis in Behçet disease: an analysis of 880 patients. Am J Ophthalmol. 2004;138(3):373–380.
Verity DH, Wallace GR, Vaughan RW, et al. Behçet’s disease: from Hippocrates to the third millennium. Br J Ophthalmol. 2003;87:1175–1183.
Table 7-3. Diagnostic Criteria for Behçet Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


