CHAPTER 200 Pediatric Head and Neck Malignancies
Each year in the United States, cancer is diagnosed in 1 of every 7000 children 0 to 14 years of age. In 1998, the diagnosis of cancer was given to 8700 children before their 15th birthday and to 3700 young adults between 15 and 20 years of age in the United States.1 Data collected by the Surveillance, Epidemiology, and End Results (SEER) Program of the National Cancer Institute (NCI) between 1972 and 1990 suggested that these numbers were on the rise. In 1972, 12 cancers were diagnosed per 100,000 children under the age of 15 years; by 1990, that number had increased to nearly 14 per 100,000.2,3 However, the rates have been stable since 1990.4 Among malignancies of the head and neck, the incidence rate increased from 1.1 to 1.49 per 100,000 person–years.5 During this same period, cancer-related mortality rate decreased. Therefore, the changes in incidence probably coincided with earlier diagnosis because of advances in central nervous system (CNS) diagnostic imaging such as computed tomography (CT) scanning and magnetic resonance imaging (MRI), and not a true increase in rates of disease.4 However, malignancies remain the most common cause of death from disease in the 1- to 14-year-old age group. Childhood cancers are exceeded only by accidents as the leading cause of death in children older than 1 year of age.
Impressive gains have been made in survival rates for some childhood cancers. In children younger than 15 years of age who are diagnosed with solid tumors, the survival rate has more than doubled—from 28% in 1960 to a more than 75% 5-year survival rate in 1993 and an 80% 3-year survival rate in 1995.6 Particularly impressive survival gains have been achieved with Wilms’ tumor and Hodgkin’s lymphoma, both of which now have cure rates exceeding 90%.2 Outcome data during this 30-year period show that the slopes of curves describing survival over time have remained fairly constant for all types of tumors. Advances in treatment and gains in survival have been achieved in a slow, incremental manner based on results of multicenter clinical trials. In spite of these advances, approximately 25% of children diagnosed with cancer will die of their disease.
The progress in survival rates for childhood cancer largely reflects the impact of clinical trials. Initiated first in 1948 to enable Farber and colleagues7 to document the efficacy of methotrexate in inducing remission in some children with acute lymphocytic leukemia, most clinical trials today are conducted by the Children’s Cancer Group or the Pediatric Oncology Group. In these trials, standard therapy is compared with a modification of that therapy, and if the modified protocol is superior, it then becomes the new standard. Progress is tedious, but from the perspective of time, it has been tremendously rewarding.
The possibility of more rapid advances in diagnosis and management is becoming increasingly real as studies of the molecular biology of childhood malignancies elucidate several requisite steps for tumorigenesis. Two important cytogenetic events, translocation and amplification, are important in tumor development and can act synergistically to generate potent oncogenic activity.8 Nonrandom chromosomal translocations typically activate oncogenes by one of two mechanisms.9 The first mechanism, frequently found in lymphoid malignancies, places the coding sequence of one gene under the regulatory control of a second gene. This juxtaposition results in increased or inappropriate expression of the first gene. The second mechanism generates a new gene, which is a chimera of two other genes that is formed by joining their coding regions. For amplification events, additional copies of a cellular gene are generated in the form of double-minute chromosomes or homogeneously staining regions.10
The pathologic evaluation of childhood tumors is very different from that of adult tumors. Because of the large numbers of mesenchymal tumors that can look very primitive and similar on light microscopy—small round blue cell (SRBC) tumors—use of newer techniques such as immunochemical, molecular, or ultrastructural analysis is necessary for accurate diagnosis. Proper diagnosis is crucial to developing a treatment plan and for prediction of clinical behavior. Every child with suspected leukemia-lymphoma or neuroblastoma should have a workup to evaluate gene translocations, antigen expression, and gene amplification. Critical to this pathologic workup is the delivery of fresh tissue to the pathologist for proper handling. Figure 200-1 shows proper tissue handling to ensure the availability of tissue for all types of analysis. Fresh tissue has the messenger ribonucleic acid (mRNA) necessary to carry out cell cultures for molecular genetic analysis and for growth of a permanent cell line. Because tissue often is not handled properly and is fixed in formalin before arriving at the pathologist’s laboratory, techniques have been developed to obtain molecular genetic information from fixed or frozen tissue by such means as reverse transcriptase–polymerase chain reaction (RT-PCR) analysis, flow cytometry, fluorescence-activated cell sorting (FACS), and fluorescence in situ hybridization (FISH). However, RT-PCR analysis requires intact RNA, which can be obtained only by snap-frozen tissue stored at −70° C from viable tumor areas without tumor necrosis. Some success has been achieved with use of fixed, paraffin-embedded tissue for RT-PCR assays, but further work is needed in this effort.
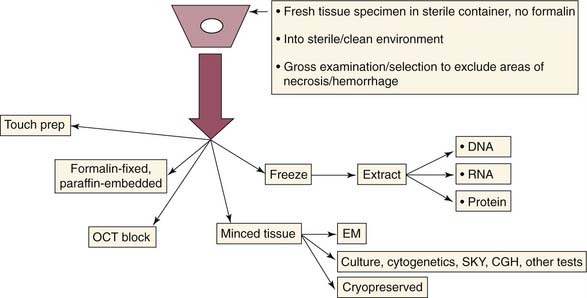
Because resources are limited, a practical way to handle biopsy tissue must be adopted (see Fig. 200-1). After delivery of fresh tissue to the pathologist, the tissue is examined by routine hematoxylin and eosin (H&E) stains. If the diagnosis is not clear, additional methods are brought into play to establish a diagnosis such as immunohistochemistry, molecular genetic analysis, electron microscopy, cytogenetic studies, and deoxyribonucleic acid (DNA) sequencing (Table 200-1). These evolving techniques are used judiciously and are rarely all needed. The most common secondary procedure after examination of H&E stains is immunohistochemistry, followed by special stains, electron microscopy, and molecular studies. Immunohistochemistry is used to confirm the histogenesis of a tumor. Approximately 35 antibodies are commonly used in pediatric tumor workups in the categories of general (vimentin), hematopoietic, neural, myogenic, epithelial, and miscellaneous. Immunohistochemistry and electron microscopy have largely replaced the use of special stains, but a few are still useful, such as trichrome, pentachrome, reticulin, myeloperoxidase, and periodic acid–Schiff (PAS) stains. Electron microscopy is used primarily to clarify conflicting results from other studies such as immunohistochemistry. Molecular genetic techniques include five major methods: polymerase chain reaction (PCR) analysis, cytogenetic studies, FISH, spectral karyotyping (SKY), and comparative genomic hybridization (CGH). These techniques are used to detect genomic alterations (translocations, amplifications, or deletions) from chromosomal analysis. Many protocols require the use of special studies for ploidy and molecular genetics to enter a patient into a protocol treatment arm. The availability of molecular genetics has revolutionized childhood cancer diagnosis, providing accurate diagnoses that help guide therapy and predict clinical aggressiveness. Molecular genetic analysis of childhood tumors should not be taken with blind faith because two problems can be encountered: certain tumor-specific translocations only occur in a percentage of tumors, and in some cases, the same translocation can occur in a phenotypically different tumor. Still, molecular genetics has enormous value in the diagnostic workup of the difficult, primitive pediatric tumors.
Table 200-1 Diagnostic Methods for Tumor Diagnosis
| Method | Comment |
|---|---|
| Light microscopy | Used in all cases |
| Immunohistochemistry | Widely used ancillary diagnostic |
| Molecular genetic: reverse transcriptase–polymerase chain reaction (RT-PCR) | Common molecular diagnostic method |
| Molecular genetic: FISH | Replacing cytogenetics in many cases |
| Molecular genetic: in situ hybridization | Specialized use |
| Special stains | Still useful in some cases, inexpensive |
| Electron microscopy | Used to augment light microscopy |
| Cytogenetics | Used when no suitable FISH probes available |
| Molecular genetic: SKY, CGH | Newer diagnostic methods |
| Molecular genetic: DNA sequencing | For rare cases |
CGH, comparative genomic hybridization; FISH, fluorescence in situ hybridization; SKY, spectral karyotyping.
Head and neck tumors of childhood are reviewed in this chapter. This subset of malignancies accounts for approximately 12% of all childhood cancers and, in order of relative prevalence, includes lymphomas (27%: Hodgkin’s lymphoma 17%, non-Hodgkin’s lymphoma 10%), neural tumors (23%: retinoblastoma and neuroblastoma), thyroid malignancies (21%, with papillary carcinoma the most common pathologic diagnosis), and soft tissue sarcomas (12%: rhabdomyosarcoma; 8%, nonrhabdomyosarcoma soft tissue sarcomas [NRSTSs]).5 Age has a marked effect on tumor type and modifies this order of prevalence considerably. The age factor is obviously pertinent to a directed evaluation and diagnostic workup (Table 200-2).

Lymphomas
Non-Hodgkin’s Lymphomas
Acute lymphoblastic leukemia, defined as leukemic involvement of greater than 25% of the bone marrow, is the most common pediatric malignancy. Lymphomas, defined as leukemic tumor cells extending beyond bone marrow sites and involving less than 25% of the bone marrow, are the third most common cancers in children in the United States,11 and non-Hodgkin’s lymphomas represent 60% of these diagnoses. The annual incidence in children younger than 15 years of age is 7 to 8 per 1 million, with peak ages between 7 and 11 years. Boys are preferentially affected in a 3 : 1 ratio,12,13 and the disease is twice as common in whites as in blacks.14 Although adult lymphomas are primarily nodal, childhood lymphomas are predominantly extranodal. Since 1973, the annual incidence of non-Hodgkin’s lymphoma has risen by approximately 30%. Although the reasons for this increase remain unclear, specific populations of children who are at greater risk for non-Hodgkin’s lymphoma have been identified. These groups include children with congenital immunodeficiency syndrome (Wiskott-Aldrich syndrome, ataxia-telangiectasia, and X-linked lymphoproliferative disease), children with acquired immunodeficiency syndrome (AIDS), and children who have received immunosuppressive therapy for organ or bone marrow transplantation. Patients treated for Hodgkin’s lymphoma and solid tumors with chemotherapy are at an increased risk for non-Hodgkin’s lymphoma as well. Common to all these patients is deficient or impaired T-cell function.
Geographic differences in the incidence rates and subtypes of non-Hodgkin’s lymphoma are striking. For example, non-Hodgkin’s lymphoma is rare in Japan, whereas in equatorial Africa and northeastern Brazil, Burkitt’s lymphoma is endemic and accounts for approximately 50% of all cancers.11 An association with Epstein-Barr virus (EBV) and BL has been demonstrated in these areas.15 Even in cases of sporadic Burkitt’s lymphoma in the United States for which standard screening yields a negative result,16 EBV genomic DNA can be detected with molecular biology.
Presentation and Evaluation
Characterized by an acute onset with rapid progression, prompt diagnosis and initiation of therapy are important to reduce morbidity and ensure long-term survival.17 In 23% to 46% of affected children, primary disease occurs within the abdominal cavity and involves, in decreasing order of frequency, the distal ileum, cecum, and appendix.18,19 Mediastinal primary tumors are slightly less common and are diagnosed in 15% to 45% of cases.20 Primary head and neck involvement occurs in 5% to 10% of children and originates in the salivary glands, larynx, paranasal sinuses, orbit, scalp, and Waldeyer’s ring.19 Proximity to the CNS makes direct extension possible.
The rapid progression of disease mandates prompt referral to a specialized management center before biopsy so that a pediatric oncologist can expedite the diagnostic and staging evaluation (Table 200-3) and determine the site and procedure to obtain tissue for histologic, cytologic, immunophenotyping, and genotyping analyses, with consultation from the surgeon and pathologist if necessary. Definitive diagnosis requires biopsy, sometimes by excision of a cervical node. Mediastinal involvement should be assessed by neck and chest CT before any procedure under general anesthesia. Disseminated disease is the rule, however, and often adequate numbers of malignant cells are found in the bone marrow, spinal fluid, or pleural or ascites fluid, obviating surgical intervention entirely. The presence of pancytopenia suggests bone marrow involvement, and when more than 25% of the marrow is replaced with tumor, children are assigned a diagnosis of acute lymphoblastic leukemia.
Table 200-3 Diagnostic and Staging Evaluation for Non-Hodgkin’s Lymphoma
| Essential | Useful | Occasionally Indicated |
|---|---|---|
| History | Intravenous urography | Contrast studies of the gastrointestinal tract |
| Physical examination | Special radiologic views (airway) | CT, MRI of the brain |
| Specimens for histology, cytology, immunophenotyping, genotyping | CT, MRI of primary site | CT, MRI of spinal cord |
| Complete blood count | Creatinine clearance | |
| Serum chemistries (electrolytes, liver, renal serum chemistries, LDH, uric acid) | Epstein-Barr virus studies | Urinary uric acid Serum lactic acid |
| Bone marrow aspiration and biopsy | Protein electrophoresis | |
| Cerebrospinal fluid analysis | Acid-base studies | Liver biopsy |
| Urinalysis | Bone scan | |
| Chest radiography | ||
| Gallium-67 scanning | ||
| Abdominal imaging |
CT, computed tomography; LDH, lactate dehydrogenase; MRI, magnetic resonance imaging.
Management
Dramatic improvement in overall cure rates among children with non-Hodgkin’s lymphoma reflects the incremental development of effective multimodal chemotherapeutic regimens through well-planned clinical trials. Systemic therapy is imperative because occult micrometastases affect more than 80% of children with non-Hodgkin’s lymphoma and cure rates are less than 20% when only local therapy is used.17 Management protocols are based on the extent of disease as determined by the staging workup and the histologic subtype.
The most widely used staging classification is the St. Jude Children’s Research Hospital classification, based on the Ann Arbor staging system for Hodgkin’s lymphoma, modified for non-Hodgkin’s lymphoma (Table 200-4). Chemotherapy with combination drug therapy is the backbone of management, traditionally with agents used for acute lymphoblastic leukemia (i.e., cyclophosphamide, doxorubicin, vincristine, and prednisone, and for extensive or recurrent disease, methotrexate, Ara-C, etoposide, and ifosfamide). Overall cure rates are in the range of 70% to 90%, depending on tumor subtype. Radiotherapy has no routine role in the treatment of non-Hodgkin’s lymphoma. Its use is reserved for massive disease, and surgery is used only occasionally in children with life-threatening tracheal compression. Response to therapy is assessed by monitoring tumor size and activity using a variety of imaging modalities or invasive techniques.
Table 200-4 Staging Systems for Non-Hodgkin’s Lymphoma
| Stage | Staging System | |
|---|---|---|
| Ann Arbor | St. Jude Children’s Research Hospital | |
| I | Involvement of a single lymph node region (I) or of a single extralymphatic organ or site (IE) | Single tumor (extranodal) or single anatomic area (nodal) excluding the mediastinum or abdomen |
| II | Involvement of two or more lymph node regions on the same side of the diaphragm (II) or localized involvement of extralymphatic organ or site and of one or more lymph node regions on the same side of the diaphragm (IIE) | Single extranodal site with regional lymph node involvement; two or more nodal areas on the same side of the diaphragm; two single extranodal tumors ± regional node disease on the same side of the diaphragm; primary gastrointestinal tract tumor, usually in the ileocecal area ± involvement of associated mesenteric nodes |
| III | Involvement of lymph node regions on both sides of the diaphragm (III) ± extranodal involvement (IIIE) ± splenic involvement (IIIS), or both (IIISE) | Two single extranodal tumors ± regional node disease on opposite sides of the diaphragm; two or more nodal areas on opposite sides of the diaphragm; all extensive primary intra-abdominal disease; unresectable; all primary intrathoracic tumors (mediastinal, pleural, thymic); all primary paraspinal or epidural tumors regardless of other sites |
| IV | Diffuse involvement of one or more extralymphatic organs; organs or tissues ± associated lymph node enlargement | Any of the above + initial CNS or bone marrow involvement (<25%) |
CNS, central nervous system.
Histopathology and Molecular Biology
The histopathology of non-Hodgkin’s lymphoma is extremely variable and has been categorized in various ways. The traditional and widely adopted Rappaport classification uses histologic pattern, cell type, and degree of differentiation to categorize tumors. In children, the categories seen with the most frequency are B-cell phenotype (Burkitt’s lymphoma, Burkitt’s-like lymphoma, large B-cell lymphoma), pre-T-cell origin (lymphoblastic lymphoma), and T-cell origin (anaplastic large-cell lymphomas and peripheral T-cell lymphomas). The diffuse histologic pattern is almost universal within these categories.17
The development of immunologic markers has added refinement to categorization and contributed to the identification of molecular mechanisms in pathogenesis. Best studied are the small, noncleaved-cell lymphomas, particularly Burkitt’s lymphoma (Fig. 200-2). In Burkitt’s lymphoma, EBV, a B-cell mitogen, probably fosters the establishment of a premalignant state by expanding the pool of lymphocytes in which chromosomal transformations can occur. Classic to Burkitt’s lymphoma is the translocation of the myc gene from chromosome 8 to a position juxtaposed to an immunoglobulin receptor subunit gene on chromosomes 14 (80%), and less commonly to chromosomes 2 or 22 [t(8;14), t(8;2), t(8;22), respectively].21,22 Unregulated expression of the myc gene disrupts regulation of the expression of target genes that are required for the normal progression of the cell cycle.23–25
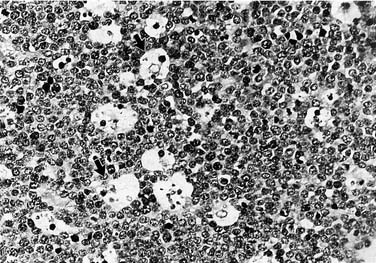
Approximately 80% of large-cell lymphomas have a t(2;5) chromosomal rearrangement.26 The precursor RNA can be detected by a RT-PCR assay, which has become an important diagnostic tool.27 Although the impact of molecular biology has yet to revolutionize clinical practice, it will inevitably refine our diagnostic capabilities and permit the development of target tumor–specific therapy.
Hodgkin’s Lymphoma
In 1832, Thomas Hodgkin described the disease that now bears his name. Although characterized by the presence of mononuclear cells (Hodgkin’s cells) and binucleated or multinucleated tumor cells (Reed-Sternberg cells) in a background of variable and often abundant reactive cells, a single unifying principle of the biologic nature of Hodgkin’s lymphoma and of the origin of the Reed-Sternberg cell has remained elusive.28 Current data suggest that Hodgkin’s lymphoma represents a group of diseases with an uncertain relationship to each other.29 Age at diagnosis follows a bimodal distribution, with the first peak occurring between 15 and 40 years of age and the second peak occurring after age 50. Approximately 4% of cases occur in children younger than 10 years of age, and 11% are found in children between 10 and 16 years of age.30
Presentation and Evaluation
In more than 90% of childhood and adult cases, Hodgkin’s lymphoma arises in the lymph nodes, typically causing an asymmetrical adenopathy that is firm, rubbery, and nontender. Particularly common is the combination of mediastinal and right supraclavicular nodal disease. The spleen is the most common extranodal site of involvement. Constitutional symptoms (B symptoms) may be present and include fever of 38° C or higher, drenching night sweats, or unexplained loss of 10% or more of body weight. Diagnosis is made by lymph node biopsy, and the disease is then staged using the classification developed in 1971 by the Ann Arbor workshop31 and modified 18 years later to incorporate newer procedures such as CT (see Table 200-4, Ann Arbor staging system).32 Substage classifications include A for asymptomatic disease, B for B symptoms (as described earlier in the chapter), and E for extension to an adjacent extranodal site. The staging system is based on the premise that Hodgkin’s lymphoma spreads through the lymphatics to contiguous nodal groups and through the circulation to extranodal sites, although these modes of spreading have not been clearly established and multifocal involvement may represent multicentric disease.29
Clinical staging procedures include history, physical examination with measurement of lymph nodes, chest radiograph, CT scan of neck and chest, CT or MRI of abdomen, and laboratory tests (complete blood count with differential, erythrocyte sedimentation rate determination, renal and hepatic function tests, alkaline phosphatase assay). CT has limited sensitivity for detection of abdominal adenopathy in children because of their lack of retroperitoneal fat.33 Therefore, abdominal MRI may be a better study for determining extent of abdominal and pelvic disease. Pathologic staging includes excisional lymph node biopsy. Bone marrow aspiration and biopsy is reserved for patients with stage III or IV disease or B symptoms. Routine use of surgical staging with splenectomy and lymph node sampling is no longer in use because of improved imaging techniques, routine use of systemic therapy, and the knowledge that splenectomy is associated with increased infectious and neoplastic complications. Stage at presentation is similar among children and adults, with approximately 60% of patients presenting with stage I or II disease and 40% of those presenting with stage II or IV disease, regardless of age.30 In children younger than 10 years of age, however, stage I disease is more common (18%) and stage IV disease is less common (3%) than in adolescents (8% and 15%, respectively) and adults (11% and 11%, respectively). Systemic B symptoms also are less likely in younger patients (19%) than in adolescents (30%) and adults (32%).
Management
Management of Hodgkin’s lymphoma varies according to stage. In most centers, radiation therapy is recommended for stage IA and stage IIA disease. Because of the high relapse rate among children with stage IIA disease of the mediastinum, chemotherapy also may be given.34 For more advanced disease, chemotherapy either alone or as part of a combined regimen including radiotherapy is used.35
Long-term results for pediatric patients with early-stage Hodgkin’s lymphoma suggest a prognosis similar to that in adults, with cure rates of approximately 90%.36 With use of combined-modality therapy for stage IV disease, overall survival rates are slightly lower (approximately 80%).35 When patients who have received only chemotherapy demonstrate relapse at nodal sites, salvage radiotherapy should be considered.37
Histopathology and Molecular Biology
The major histologic types of Hodgkin’s lymphoma recognized by the Lukes, Butler, and Rye classifications include lymphocytic predominance, mixed cellularity, nodular sclerosis, and lymphocytic depletion (Fig. 200-3).29 Further subclassification probably is unncessary, because current management protocols have eliminated differences in survival rates between the most common types (mixed cellularity and nodular sclerosis).
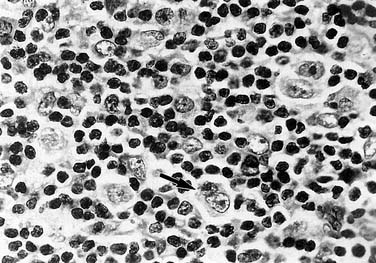
Micromanipulation of histologic sections to isolate and analyze individual Hodgkin’s and Reed-Sternberg cells has verified an origin from B-lineage cells at various stages of development.38 Ig heavy chain rearrangements have also been detected by PCR amplification in approximately 25% of cases.39 These studies suggest that Hodgkin’s lymphoma represents a clonal lymphocyte population and that different chromosomal rearrangements may be associated with different subclasses of disease.
Post-Transplantation Lymphoproliferative Disorder
As solid organ transplantation success rates have improved, diseases related to chronic immunosuppression have increased in prevalence. Post-transplantation lymphoproliferative disorder (PTLD) is the term for all abnormal proliferations of lymphoid tissue in the transplant recipient, ranging from lymphoid hyperplasia to malignant non-Hodgkin’s lymphoma. Risk factors for the development of PTLD are the degree of immunosuppression, use of potent immunosuppressive agents such as tacrolimus, EBV seronegative status at time of transplantation, young age, donor-recipient mismatch, severe graft-versus-host disease (GVHD), T-cell depletion of donor marrow, use of anti–T-cell monoclonal antibodies, and high-dose antithymocyte globulin for GVHD prophylaxis. The rate of PTLD ranges from 8% to 28%,40 depending on the immunosuppressive agents used. The rate of PTLD is higher in liver, heart, and heart-lung transplant recipients. The rate of PTLD is lower for renal transplant recipients, probably because of the higher level of immunosuppression needed in the former group.41 Most PTLDs are of B-cell origin, but early PTLD can manifest as plasmacytic hyperplasia or as a polymorphic lymphoid process.42 When PTLD affects the tonsils and adenoids, it often is of the plasmacytic hyperplasia type. When PTLD is seen in the lymph nodes, it is often a polymorphic lymphoid lesion. Treatment of PTLD includes decreasing the immunosuppression, to which most patients respond. When a transplant recipient presents with enlargement of the tonsils and adenoids, removal of the affected tissue (i.e., by tonsillectomy and adenoidectomy) is indicated to evaluate the tissue for PTLD. Removal of the affected tissue often is curative. Other therapies for PTLD include intravenous immunoglobulin, interferon alfa, antiviral therapy (with acyclovir or ganciclovir), and immunotherapy with donor T-lymphocyte infusion.
A correlation between EBV infection and PTLD is well documented.43 In immunosuppressed patients, the T-lymphocyte response to the EBV infection is suppressed and the B-cell proliferation turned on by the virus infecting the B-lymphocytes is allowed to proceed unchecked. Thus, EBV seronegativity at the time of transplantation is a risk factor for the development of PTLD, because active EBV infection after transplantation can initiate this polyclonal response from the B lymphocytes infected with the Epstein-Barr virus.
Sarcomas
Rhabdomyosarcoma
Rhabdomyosarcoma is the most common soft tissue sarcoma of childhood, representing 13% of all pediatric malignancies. Rhabdomyosarcoma is the third most common neoplasm in childhood after neuroblastoma and Wilms’ tumor. The annual incidence of this tumor in children younger than 20 years of age is 4.3 cases per 1 million children, affecting approximately 350 children in the United States each year.44 Approximately 35% of these children present with disease in the head and neck. Sites of involvement include the orbit, nose and paranasal sinuses, oropharynx, soft tissues, nasopharynx, and external ear or mastoid. Because of the high possibility of contiguous spread to the CNS, the paranasal sinuses, infratemporal fossa, middle ear, and nasopharynx are classified as parameningeal sites.
In the 1960s, fewer than one third of children with rhabdomyosarcoma survived, but cure rates now approximate 70%, largely reflecting advances made by the Intergroup Rhabdomyosarcoma Study Committee (IRSC).45,46 Before the formation of the IRSC in 1972, the relatively small numbers of patients, short follow-up times, and the confounding effects of varied treatments limited univariate and multivariate analyses, especially for small subgroups of patients. However, when three pediatric centers agreed to pool patients and investigate resources, it became possible to identify prognostic variables of crucial importance for stratifying patients in clinical trials. Valid comparisons of alternative therapies were developed, and therapies were tailored according to the risk of relapse. The data obtained through the collaborative, multimodality therapeutic protocols developed and studied by the IRSC have allowed a steady improvement in the curability of rhabdomyosarcoma, especially the locally extensive tumors.
Presentation and Evaluation
A majority of affected children are younger than 6 years of age when the diagnosis of rhabdomyosarcoma is made.47 The spectrum of presenting signs and symptoms varies greatly depending on the site of the primary tumor, the age of the child, and the presence or absence of metastatic disease. Presentation in the head and neck most frequently is heralded by a local swelling. Fewer than one half of these children have pain. Nasal discharge or obstruction, signs of cranial nerve involvement, otorrhea, hearing loss, fetor, and proptosis are even less frequent.48
The orbit is involved in nearly one third of rhabdomyosarcomas of the head and neck,48 with the typical presentation being that of a rapidly developing, unexplained proptosis. In decreasing order of frequency, rhabdomyosarcoma also affects the oral cavity and pharynx (29%), the face and neck region (24%), and the ears and sinuses (9%). Nasopharyngeal and paranasal sinus involvement can cause nasal obstruction or epistaxis, and otitis media, facial nerve palsy, or tumor filling the ear canal is often the presenting symptom of temporal bone disease.49
Surgery is a necessary part of the diagnostic and staging evaluation and is an essential component in the Intergroup Rhabdomyosarcoma Study Clinical Grouping Classification (IRSCGC) (Table 200-5). Although this staging system is widely used, it was revised in 1991 and the primary tumor, regional lymph nodes, and distant metastasis (TNM) classification is now used for Intergroup Rhabdomyosarcoma Study (IRS) patients in IRS groups IV and beyond. For instance, patients in IRSCGC group III represent a heterogeneous collection of patients who have resectable and unresectable tumors, depending on the experience of the institution. As surgery techniques have advanced, especially in the skull base, and reconstructive techniques such as microvascular reconstruction are more widely available, complete extirpation often is feasible for rhabdomyosarcoma in the head and neck.48 In some centers, however, surgery may be limited to partial removal or excisional biopsy to establish the diagnosis. Additional important variables that were not considered in the IRSCGC were the degree of local extension and the status of the primary echelon nodes. To address these shortcomings, the IRSC now uses the TNM classification, similar to that used to characterize disease extent in adults with solid tumors (Table 200-6).
Table 200-5 Intergroup Rhabdomyosarcoma Study Clinical Grouping Classification*
| Group | Definition |
|---|---|
| I | Localized disease, completely resected |
| II | Compromised or regional resections with microscopic residual disease |
| III | Incomplete resection or biopsy with gross residual disease |
| IV | Distant metastatic disease present at onset |
* Used in Intergroup Rhabdomyosarcoma Studies I through III.



