Fig. 14.1
Distal pancreatectomy specimen showing insulinomas in a MEN1 patient
14.2.4 Pathophysiology and Clinical Features
In patients with insulinoma, there is continued secretion of insulin despite low blood glucose; such break of the physiologic negative feedback with the resulting inappropriately excessive insulin secretion is the pathological hallmark of insulinoma [22]. The clinical spectrum of hypoglycemia is attributed to reduced hepatic glucose production rather than increased utilization [23]. Patients present typically at their fourth or fifth decade [24], and the diagnosis is sometimes made years after presentation [16] due to diversity and ambiguity of symptoms. Blood glucose levels below 55 mg/dl induce excessive catecholamine release and subsequently result in appearance of adrenergic symptoms like anxiety, palpitations, tremors, weakness, hunger, nausea, sweating, and warmth. Severe hypoglycemia with blood glucose level dropped below 50 mg/dl causes neuroglycopenia manifested as neurologic dysfunction in the form of visual disturbances (diplopia, blurred vision), amnesia, confusion, convulsions, and even coma [13, 16, 22, 23].
Two points should be emphasized while clinically evaluating these patients. First, timing of symptoms should be confirmed; early morning or post-exercise onset usually favors the diagnosis of insulinoma, while postprandial symptoms, though cannot preclude [15], yet do not favor its diagnosis [13]. Second, given the rarity of the condition and the usually nonspecific presenting symptoms, clinical suspicion should always be kept high.
14.2.5 Biochemical Diagnosis
When clinically suspected, diagnosis is usually suggested by detecting hyperinsulinemia in the presence of hypoglycemia plus reversal of the symptoms by administration of glucose, fulfilling classical criteria of Whipple triad [22]. The current consensus [15] to confirm diagnosis depends on demonstration of six criteria during 72-h fasting test, though some studies declared the adequacy of 48-h fasting [25]. These criteria are (1) documented blood glucose levels ≤2.2 mmol/l (≤40 mg/dl), (2) concomitant insulin levels ≥6 μU/ml (≥36 pmol/l; ≥3 U/l by ICMA), (3) C-peptide levels ≥200 pmol/l, (4) proinsulin levels ≥ 5 pmol/l, (5) β hydroxybutyrate levels ≤2.7 mmol/l, and (6) absence of sulfonylurea (metabolites) in the plasma and/or urine.
14.2.6 Imaging Localization
Once the diagnosis is biochemically confirmed, localization is sought. Various imaging modalities are available with variable sensitivities. None of them is solely adequate, so more than single modality may be used; however, localization can be accomplished preoperatively in the vast majority of cases. Initially, anatomic imaging modalities are used. Computed tomography (CT) is generally preferred over transabdominal sonography because of reported higher sensitivity in most [26–28] – but not all [24] – studies, in addition to the traditional limitations of ultrasound (US) in obese patients and gaseous abdomen, which are easily overcome by CT scan (Figs. 14.2 and 14.3). Moreover, technical advances in CT enhanced its sensitivity enabling localization of more than 94 % of insulinomas [29]. If the tumor is yet not localized, endoscopic ultrasound (EUS) is the appropriate next step. The disadvantages of EUS, being invasive and requiring expertise, are usually overlooked in light of its very high sensitivity which reached 100 % in some studies [26]; somatostatin receptor scintigraphy (SRS), as a sole modality, has low sensitivity for the detection of insulinoma owing to the usually small tumor size and low density of somatostatin receptors in the tumor [30].
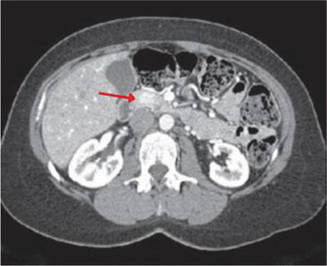
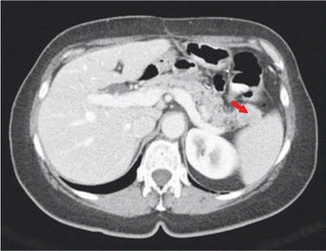
Fig. 14.2
CT scan demonstrating a 1.4-cm hypervascular mass in the head of the pancreas in a patient diagnosed with insulinoma (arrow)
Fig. 14.3
CT scan demonstrating a tumor in the tail of the pancreas (arrow)
For the remaining minority of cases, functional imaging is resorted to. Angiography, arterial stimulation venous sampling (ASVS), or selective arterial calcium stimulation (SACS) helps to localize the tumor by verifying hormonal function and thus allowing a more accurate surgical approach [15].
Magnetic resonance imaging (MRI) is an alternative to CT with a higher sensitivity, 50 % versus 30 %, in some studies [31]. However, being much more expensive, it is reserved for patients in whom contrast is contraindicated. Occult insulinomas refer to a minority of tumors which are biochemically confirmed yet cannot be preoperatively localized despite the use of the aforementioned localization studies [11].
14.2.7 Management
In managing an insulinoma patient, two goals should be approached, hypoglycemia control and oncological control, and in fact it is ideal to achieve the first via approaching the second. In addition, given the fact that insulinomas – in the vast majority of cases – are benign and solitary, surgery is usually curative and therefore is indicated. Medical treatment, however, is indicated for preoperative control, for unresectable disease because of metastasis, and for the patient unwilling or unfit for surgery [7]. In order to ameliorate hypoglycemic symptoms in these cases, the following nonoperative strategies may be attempted: (1) dietary modification in the form of slowly absorbed carbohydrates and more frequent meals. Continuous glucose infusion may be needed in some patients with severe hypoglycemia. (2) Antihypertensive agent diazoxide may be used on account of its hyperglycemic potential through a dual action and pancreatic through inhibition of insulin release and extrapancreatic through potentiating glycogenolysis. However, its use may be associated with major adverse reactions like nausea, sodium retention with edema, weight gain, and hirsutism [11]. As previously mentioned, surgery is the only curative treatment for insulinomas. While continuously and carefully monitoring blood glucose level, the tumor is approached through a bilateral subcostal incision followed by complete pancreatic mobilization. An initial exploration is done to exclude metastatic disease. Meticulous examination of the entire pancreas, both bimanually and by intraoperative US, is done to identify the tumor(s) and its (their) relation to major pancreatic ducts (Fig. 14.4). Enucleation is usually done, though distal pancreatectomy or rarely pancreaticoduodenectomy may be performed as indicated by tumor location and size. Blind resection and progressive resections are prohibited. Drainage is generally recommended [13]. With the advent of endoscopic instruments and vascular sealing devices, laparoscopic resection has also been successfully performed [22] albeit with a higher incidence of pancreatic fistulas [23]. Postoperatively, some patients, being transiently hyperglycemic, may require small doses of insulin, avoiding glucose in the first 24-h IV fluids. Drains are removed when the patient resumes oral intake and pancreatic leakage is not suspected [23].
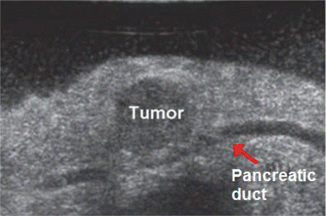
Fig. 14.4
Intraoperative US demonstrates the close proximity of the pancreatic duct (PD) just deep to the tumor
In their series, Hirshberg et al. [32] reported a prolonged survival in nine out of ten patients with malignant insulinoma after appropriate surgical resection. In addition, Starke and colleagues reported the results of follow-up in ten patients with insulinoma and liver metastasis who underwent successful pancreatic surgery and reported the death of four patients from unmanageable hypoglycemia [33]. These reports explain the aggressive surgical approach justified even in metastatic cases. Peripancreatic lymphadenectomy should be performed in case of suspected or proven malignancy [13].
Resection of hepatic metastases, synchronous with the appropriate pancreatic surgery, has been recommended [34]. For patients with insulinoma-induced intractable hypoglycemia, that is not amenable for surgery, continuous glucose infusion pump has been used [16]. An induced apoptotic and antiproliferative effect was reported via targeting glucagon-like peptide-1 receptors on insulinoma cells; this may provide future novel therapy for inoperable cases [35].
14.3 Gastrinoma
14.3.1 Historical and Epidemiological Aspects
Gatrinomas were first described as a distinct clinicopathological entity by Zollinger and Ellison in 1955 [36]. They occur at an annual incidence of 0.5–2/million of population and are the most common functional malignant P-NET [7] and are second in incidence to insulinoma among gastropancreatic NETs associated with hormonal syndrome. They are most frequently located in the duodenum and are the sole functioning duodenal NET [37]. Their clinical importance originates primarily from their ulcerogenic potential and malignant potential [38]. Although 80 % of cases are sporadic, 20 % are familial, occurring in the setting of MEN1 syndrome [39]. While many aspects of the disease are established facts, its management – particularly advanced and familial cases – is a subject of considerable debate.
14.3.2 Genetic Basis
Numerous genetic abnormalities have been identified to be involved in the pathogenesis of gastrinoma. A germline mutation of the tumor suppressor gene MEN1 located on 11q31 was found not only in familial cases [40] but also in sporadic ones [41]. A number of chromosomal deletions, 1q and 22q, have also been identified in 44 and 96 % of gastrinomas, respectively, and may be associated with aggressive growth pattern and higher metastatic rates [42, 43]. A number of tyrosine kinase receptors seem to be also involved. In a small series by Evers, amplification of the HER-2/neu gene was detected in all gastrinoma samples [44]. Similarly, insulin-like growth factors (IGF1 and IGF 2) are expressed in almost all gastrinomas [45].
14.3.3 Pathological Features
Gastrinomas arise from neuroendocrine cells and are well-differentiated tumors. Nevertheless, more than 60 % of them are malignant based on the biologic behavior rather than on histology [13]. The site of the tumor has no effect on the age of diagnosis, nor the severity of disease, but may minimally influence the clinical presentation [46]. The majority of gastrinomas are located in the duodenum, pancreatic head, and lymph nodes within the gastrinoma triangle whose heads are cystic–common bile duct junction, pancreatic head–body junction, and duodenal second–third parts junction. Synchronous occurrence of pancreatic and duodenal gastrinomas was reported in 11 % of gastrinoma cases [38] and in 6 % of all NETs [47]. Ectopic gastrinoma, defined as extrapancreatic, extraduodenal, and extralymphatic primary gastrinoma, has been reported. In 1997, Wu et al. reviewed 20 reported cases of this rare entity and concluded that such ectopic sites should be carefully examined if no gastrinoma was found in the usual locations [48]. These ectopic sites include the stomach, jejunum, lesser omentum, liver, right kidney, and ovary. Multiplicity and associated islet cell hyperplasia are as well documented, not only in most patients with MEN1[49] but also in up to 15 % of sporadic gastrinoma [47]. In its sporadic type, patients tend to have an isolated duodenal or pancreatic gastrinoma [37]. Although those located in the duodenum are typically small (<1 cm), regional nodal and hepatic metastasis is present in about 60–80 % and 10 % of cases, respectively [50]. The proliferative activity of these tumors ranges from 2 to 10 %, and some may show angioinvasion [37]. On the contrary, pancreatic tumors are larger (>2 cm) with similar incidence of nodal involvement but higher likelihood of hepatic metastasis [51], higher proliferative activity, and higher angioinvasion rate [37]; this explains why pancreatic tumors are more malignant than duodenal ones. On the other hand, almost all gastrinomas, in MEN1 settings, reside in the duodenum, usually small (<1 cm) and, contrary to the sporadic tumors, are multicentric with distant metastasis rarely detected at initial diagnosis [52]. Finally, all gastrinomas are expressing receptors for somatostatin [53].
14.3.4 Pathophysiology and Clinical Features
Overproduction of gastrin by these tumors causes gastric acid hypersecretion which leads to peptic ulcer disease or gastroesophageal reflux disease which are in turn responsible for most clinical features of the disease spectrum known as Zollinger–Ellison syndrome (ZES).
Among 355 gastrinoma patients recorded by Soga et al., seven were asymptomatic. Asymptomatic cases were more likely to be extrapancreatic [54]. The mean age at onset of symptoms is 48–55 years in sporadic form, with slight male predominance (54–56 %). However, the familial type usually presents earlier (mean 32–35 years) [55]. In a series by Roy et al. [46], abdominal pain and diarrhea were the most frequent symptoms occurring in about three-fourths of patients, though pain was less frequently reported in familial cases and diarrhea was more frequently reported in metastatic cases [54]. Heartburn and weight loss were less common presentations, occurring in 44 % and 15 % of patients, respectively, while one-fourth of patients presented with gastrointestinal bleeding [46], melena being more common than hematemesis [54]. Other features are nausea and vomiting occurring in 12–30 % of cases and steatorrhea in 5–10 % [56]. Abdominal tumor and hepatomegaly were also reported, both being significantly more common in metastatic cases [54].
14.3.5 Biochemical Diagnosis
Biochemical assays and imaging are the mainstay of diagnosis since physical examination is usually nonspecific. The diagnosis of ZES is based on the detection of elevated serum gastrin in a fasting patient with raised gastric acid secretion. In this context several points are to be considered. Firstly, patients with pernicious anemia, atrophic gastritis, and those with medically or surgically suppressed gastric acid secretion have elevated serum gastrin level, hence the importance of demonstrating hyperchlorhydria for establishing the diagnosis of ZES and the importance of discontinuing antisecretory medications at least 7 days before the test. Secondly, criteria of basal acid output value more than 15 mEq/h or PH less than 2 are used to define the hyperchlorhydria state [57]. Thirdly, serum gastrin level required to diagnose ZES is a subject of discussion in the literature; this was defined by Harvey and Berber as more than ten times the upper limit of normal [13], while Roy et al. considered a level more than 150 pg/ml [57]. In their large prospective cohort, Berna et al. [58], however, reported normal FSG in up to 3 % of patients in addition to the finding that nearly half of the patients had FSG less than ten times upper normal, a level that overlaps with more common condition like Helicobacter pylori infection and antral G-cell hyperplasia. In case of normal FSG despite clinical suspicion or in case of equivocally elevated FSG, a provocative test is indicated in which secretin, at a dose of 2 mg/kg, is administered intravenously. In gastrinoma patients, serum gastrin level rises above baseline, in 94 % of patients, within three minutes after injection with 100 pg/ml [59]. An alternative but less sensitive provocative test is calcium infusion which, in 84 % of cases of gastrinoma, would result in 50 % rise in serum gastrin above baseline [59].
14.3.6 Imaging Localization
Once diagnosis is biochemically established, imaging localization is the next step. Two types of imaging studies are used for localization: anatomic and functional. In a study by Kisker, sensitivities of the three anatomic imaging modalities were reported to be 44 % for ultrasound, 56 % for CT, and 25 % for MRI [60]. In 1999, Norton et al. [61] found that US, CT, MRI, and angiography localized 24, 39, 46, and 48 % of sporadic gastrinomas. They also noticed that the latter three modalities achieved significantly higher sensitivities for localizing MEN1-associated gastrinomas (70 %, 71 %, and 85 %, respectively). Gibril et al. [62] studied 24 gastrinoma patients with proven liver metastasis, and they reported sensitivities of 46, 42, 71, and 62 for the detection of the metastatic disease by US, CT, MRI, and angiography, respectively. More advanced anatomic imaging like contrast-enhanced spiral CT and high-resolution MRI have been used for MEN1-associated gastrinomas (Fig. 14.5), though their sensitivity for detecting small duodenal tumors is rather low [63]. Collectively, 50–72 % of pancreatic gastrinomas were localized using anatomic imaging, which unfortunately failed to localize 80 % of duodenal tumors [13].
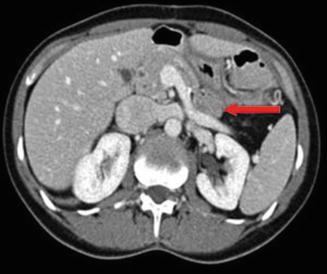
Fig. 14.5
CT scan of the pancreas with intravenous contrast. The arrow indicates gastrinoma in the pancreatic tail. The patient had MEN1 and two additional tumors of the pancreas (body and head, <1 cm each)
Knowing that almost all gastrinomas express somatostatin receptors on their cells, SRS using somatostatin analogue pentetreotide labeled with a radioactive material – indium111 – is being used for the functional imaging of these tumors. SRS was reported to have sensitivity ranging from 58 to 80 % for localization of the primary tumor [13] and 92 % for the detection of metastatic liver disease [63]. Additionally, it detected bone metastasis in all patients [64]. Gibril et al. [62], thus, concluded that SRS, given its sensitivity and cost-effectiveness, should be the initial imaging modality requested for patients with ZES. However, its sensitivity is significantly lower in smaller tumors [65]. In addition, SRS was unable to detect one-third of primary tumors subsequently detected at surgery. Alexander and colleagues [66], therefore, concluded that negative results of SRS in localizing extrahepatic gastrinomas should not be used to decide operability. The EUS was 93 % sensitive for the localization of pancreatic tumors with an average diameter of 1.51 cm [67] though its sensitivity for localizing duodenal tumors was only 40 % [68].
For patients in whom noninvasive modalities prove unable to localize tumor, invasive methods are usually needed. In a prospective study by Sug et al. [69], portal venous sampling was 71 % sensitive, while selective arterial secretin injection was 96 % sensitive for localizing gastrinomas. In a review by Fendrich et al. [63], surgical exploration with intraoperative US was concluded to be the best localizing method.
14.3.7 Management
In managing gastrinoma patients, two goals are to be approached, namely, acid hypersecretion control and oncological control. Control of acid hypersecretion must be accomplished acutely and for long term in order to protect against complications of peptic ulcer disease [70]. Historically, surgery, either vagotomy or gastrectomy, was used. However, control is now commonly adequately achieved medically via antisecretory medications, H2 blockers, and proton pump inhibitors (PPIs). Generally, PPIs are preferred over H2 blockers because their longer duration of action allows less frequent dosing conferring better compliance, in addition to their better safety profile even in case of long-term use. Omeprazole 40 mg twice daily is commonly prescribed. Up-titration may be indicated in complicated cases like MEN1 with hypercalcemia [49] and severe gastroesophageal reflux disease (GERD), while down titration has been recommended following initial control and endoscopic documentation of ulcer healing [71].
Medical control is, however, not without cons; long-term use of acid suppression medication was proved to be associated with vitamin B12 deficiency [72]. Additionally, two important issues are not addressed through acid-suppressing medications. Firstly, chronic hypergastrinemia with its well-documented predisposition to gastric carcinoids [73] and secondly, the tumor itself, which may prove malignant and may even metastasize. Thus, surgery is the only treatment that can cure gastrinoma [55].
A distinction between sporadic and familial cases is essential to draw an appropriate management plan. For sporadic cases, role of surgery is well established. In a prospective study conducted by Norton et al. [74], they compared survival rates between operated (n = 160) and nonoperated (n = 35) patients with sporadic gastrinomas and concluded that routine surgical exploration increases survival among ZES patients. Therefore, in the absence of diffuse liver metastatic disease and in the presence of acceptable surgical risk, surgical exploration is routinely recommended for all patients with sporadic gastrinoma [61].
For pancreatic lesions, simple enucleation may be safely performed for solitary well-defined lesion; distal pancreatectomy is indicated for tail lesions, while pylorus-preserving pancreaticoduodenectomy (PPP) is justified for large head lesions [63]. Duodenotomy is routinely indicated while exploring ZES patients [75]. Identified duodenal gastrinomas are subsequently enucleated if smaller than 5 mm or excised with full thickness of the duodenal wall if larger. This is followed by longitudinal suturing of the duodenal opening [63].
For MEN1-associated gastrinoma, the role of surgery is controversial due to multiplicity of the tumor, rarity of cure even after surgery, and the possibility of hepatic metastasis even in surgically managed ZES patients [76]. Firstly, whether or not to operate? Stemming from the fact that surgery never cures ZES, Mignon and Cadiot recommended conservative approach [77]. On the other hand, surgery is recommended by others based on the fact that surgery reduces the incidence of hepatic metastasis [76]. Secondly, when to operate? A selective approach recommending surgery for large-sized tumors was proposed as these were found to be associated with a higher risk of liver metastasis [78, 79]. A more aggressive approach advocating surgery for all patients with sure diagnosis of gastrinomas has also been proposed by other authors [80]. Another controversy exists concerning the management of metastatic gastrinoma. Given the facts that PPIs are extremely effective for controlling symptoms of gastric acid hypersecretion and low rate of prolonged survival reported in cases of gastrinoma with liver metastasis [20], such patients are generally not recommended for surgery. On the other hand, House et al. [81] reported a survival advantage in their series conferred by concurrent hepatic and pancreatic resection in patients with pancreatic islet cell tumors having liver metastasis. Furthermore, acceptable morbidity and mortality were reported with aggressive resection of advanced P-NETs [82]. Consequently, aggressive surgical approach for managing such cases has also been suggested.
14.4 Glucagonoma
14.4.1 Historical and Epidemiological Aspects
A clinical spectrum referring to glucagonoma was described in 1942 by Becker et al. [83]. However, it is not until 1966 when an elevated glucagon level was documented in a similar clinical case by McGavran and colleagues [84]. Glucagonoma is an RF-PNET occurring at an annual incidence of 1 per 20 million of population ranking the third after insulinoma and gastrinoma among functional P-NETs [85] arising from alpha cells of islets of Langerhans in the pancreas and expectedly, from its name and origin, produce an excess of hormone glucagon. Approximately, 60 % of cases are sporadic and the remainder occurs in the setting of MEN1 [86].
14.4.2 Pathological Features
Glucagonomas are generally solitary and large sized (more than 4 cm) [13], located in the pancreas in more than 95 % of cases, and malignant in 50–80 % of cases [6] with higher malignancy rates in larger tumors [87] and lower malignancy rates in MEN1-associated tumors [88]. Metastasis is present in about 90 % of cases, the liver being the most common site with two-thirds of cases having multiple foci usually involving both lobes [89].
14.4.3 Pathophysiology and Clinical Features
Glucagonoma patients characteristically present clinically with 4Ds syndrome: diabetes, dermatitis (Fig. 14.6), deep venous thrombosis, and depression. Other features include cheilitis, glossitis (Fig. 14.7), anemia, weight loss, and hypoaminoacidemia [20].
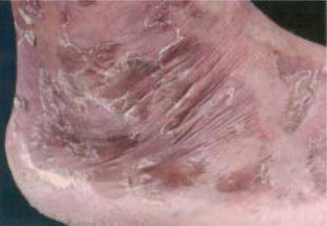
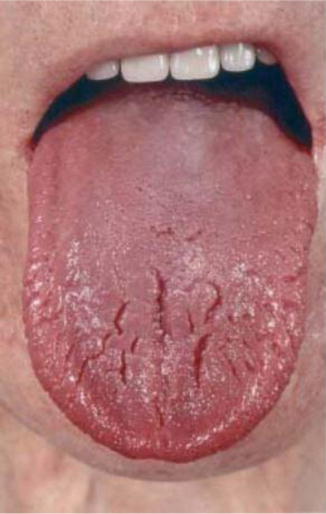
Fig. 14.6
Typical dermatitis in a patient with glucagonoma
Fig. 14.7
Typical glossitis in a patient with glucagonoma
The diabetic state is a consequence of the glycogenolytic and gluconeogenic effects of glucagon [90]. It develops in about 75 % of patients [89] and is usually mild and effectively controlled with oral hypoglycemic agents, though insulin may sometimes be required. Complications of diabetes do not occur though few cases of diabetic ketoacidosis (DKA) were reported [91, 92].
Dermatitis or necrolytic migratory erythema is a pathognomonic feature occurring in up to 70 % of patients [20], and it may be the presenting sign of glucagonoma [93]. It usually starts in the perigenital, perineal, and perioral areas and subsequently spreads centrifugally to the distal extremities [94], passing through multiple stages: erythema to vesiculation, to crusting, and to resolution over 1–2 weeks [2].
Although the exact pathophysiology has not yet been certainly elucidated, several mechanisms have been postulated, namely, hyperglucagonemia with increased protein catabolism [95], hypoaminoacidemia [86], hypoalbuminemia [90], essential fatty acid, zinc [89], and vitamin B deficiencies [94]. Amino acid hyperalimentation appears to clear this rash [86]. Deep vein thrombosis (DVT) occurs in up to 30 % of glucagonoma patients [20] and is caused by production of a molecule similar to coagulation factor X by tumor cells [90]. Weight loss is present in 62 % of symptomatic patients, may be marked, often associated with anorexia [89], and is attributed to catabolic effect of the chronically elevated glucagon level [86]. Hypoaminoacidemia is due to increased protein degradation stimulated by excess glucagon [85] and usually affecting glucogenic rather than branched-chain amino acid [87]. Anemia may occur in variable proportion of patients, normocytic normochromic in type, and caused by direct suppression of bone marrow by glucagon [89].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


