Chapter 37 Neuroprotection
History and definitions
Retinal ganglion cell glaucomatous disease
Glaucoma is a degenerative optic neuropathy characterized by loss of RGCs and their axons and warrants consideration under retinal neuroprotection strategies. Elevated IOP is a principal risk factor. However, recent evidence indicates that IOP control does not prevent glaucomatous progression in all patients and that vision loss continues in some susceptible individuals despite lowering the IOP. RGCs are terminally differentiated neurons and cannot regenerate by cell division. Optic nerve damage is irreversible. The main goal of neuroprotection is to keep these cells alive to augment the therapeutic benefit achieved by lowering the IOP. Neuroprotection in glaucoma offers a means to prevent the irreversible loss of RGC and ideally should be efficacious irrespective of the specific pathophysiology of the disease.1–3
Three decades of work have established the causal relationship between elevated IOP and the development of optic nerve damage through a series of clinical treatment studies supported by the National Eye Institute (NEI), including the Advanced Glaucoma Intervention Study trial, the Collaborative Initial Glaucoma Treatment Study Trial,4 and the Early Manifest Glaucoma Trial.5 However, glaucomatous optic neuropathy can occur despite having ocular pressures within a normal range. In some populations only half of glaucoma cases involve IOP elevated beyond the traditional demarcation of 20 mmHg pressure.6 Even in this condition of “low-tension glaucoma,” IOP remains a risk factor, as lowering the IOP further retards the loss of vision and presumptive retention of ganglion cells as monitored through nerve fiber volume in the optic nerve cup. This was shown in the Collaborative Normal Tension Glaucoma Study.7 Nevertheless, a number of individuals continue to suffer progressive glaucomatous optic nerve damage and vision impairment despite appropriate regulation of IOPs. It is these cases in particular that would benefit from an alternate strategy of neuroprotection, aimed at preventing RGC death.
Among the first studies to demonstrate pharmacologic modulation of neural damage in glaucoma was that of Bernard Becker and colleagues using oral administration of diphenylhydantoin (phenytoin).8 Diphenylhydantoin is an antiepileptic agent for controlling seizures but without the sedative effects of phenobarbital. It stabilizes the inactive state of voltage-gated sodium channels which could modulate glutamatergic transmission, and it is neuroprotective in a rat model with elevated IOP from cautery of episcleral veins.9 Diphenylhydantoin has a wide range of actions on neurons and primarily involves cell excitability, and the potential protective action on RGCs could be through ameliorating excitotoxic N-methyl-d-aspartate (NMDA) action on third-order retinal neurons. While diphenylhydantoin has numerous neurologic effects and has been tried as a central nervous system (CNS) neuroprotective agent in a number of clinical trials, it has consistently failed to meet clinical endpoints in trial.
Two neuroactive drugs that have antiexcitotoxicity action currently have Food and Drug Administration approval for CNS degenerative conditions, although clinical efficacy is limited. Riluzole extends life expectancy for amyotrophic lateral sclerosis patients by up to 3 months, and memantine is approved for advanced Alzheimer disease and provides some improvement in cognition and behavior.10 Riluzole blocks glutamatergic neurotransmission in the CNS by inhibiting release of glutamic acid, possibly through inactivation of voltage-dependent sodium channels.11 It may act at presynaptic NMDA receptors.12
Memantine is an antagonist at NMDA receptors13 and was originally developed as a dopaminergic compound derivative from amantadine, which has approval for treatment of Parkinson disease. Memantine has therapeutic benefit against excitoneurotoxicity mediated by action of NMDA receptors. A broad-spectrum blockade of NMDA channels would not be physiologically attractive. However, memantine does not compete with glutamate for the binding site. Rather, memantine binds only to open-state NMDA channels. It does not impede neuronal synaptic signal transmission in the physiological range, as it is competitive only for high levels of glutamate that lead to neuronal damage and death through excitotoxic mechanisms caused by excessive activation of NMDA receptors. The drug is well tolerated in treatment of Alzheimer dementia without apparent side-effects, suggesting that, when applied to physiologic neuronal states, it does not impede neuronal activity. Studies indicate that memantine decreases the level of free glutamate in the vitreous of a dog glaucoma disease model14 and in monkey and human glaucoma,15 although some of this work is not fully accepted. This provides a basis for considering NMDA receptor antagonists in a neuroprotection strategy for glaucoma.
Large-scale formal studies of memantine have been conducted involving patients with chronic glaucoma. In 2000–2006 Allergan ran a phase III randomized, placebo-controlled trial to evaluate the efficacy of memantine in 1179 patients aged 18–82 years old.4 Entry criteria required evidence of glaucomatous damage by examination of the optic nerve head accompanied by visual field loss. Subjects with all forms of open-angle and chronic angle closure glaucoma were entered, including having prior glaucoma filtration surgery. Two treatment arms had different doses of memantine versus an oral placebo for glaucoma patients with otherwise controlled IOP. Visual fields were monitored every 6 months for evidence of progression as the outcome measure. Unfortunately, the trial failed to meet endpoint significance versus placebo control. As the outcome has not been formally submitted for publication, information on the outcome is limited, but the memantine trials demonstrated statistically significant visual field benefit under high dose conditions compared to lower dose, although the overall trial significance failed at efficacy compared to control.4 Precise reasons for failure are not understood, but it may have had to do with study design and handling of data acquisition at the point that a subject reached the clinical endpoint. The medical glaucoma field, nonetheless, remains optimistic that the strategy of neuroprotection for RGC degenerative disease remains important for study and will ultimately yield compounds that can augment lowering of IOP in glaucoma.1,5
Neurotrophic factors in glaucoma
Recent studies suggest that neurotrophic factors may have therapeutic benefit for individuals with advanced glaucoma. Lambiase and colleagues2 demonstrated that application of nerve growth factor (NGF) in glaucomatous rats protects the RGCs from apoptotic cell death. Rats were made glaucomatous through injection of hypertonic saline into the episcleral vein, and NGF was applied topically in one of two doses (100 and 200 µg/mL). Cell biology studies indicated that molecules upstream of the apoptotic pathway, including Bax and BCL-2, were lower in treated animals, and morphometric analysis showed a significant preservation of RGC counts in the treated eyes.
These investigators then initiated a pilot human study of 3 patients with advanced glaucoma, who underwent careful electrophysiologic and psychophysical visual testing at baseline and after topical NGF eye drops applied four times daily for 3 months.2 The subjects were evaluated by multiple parameters, including visual acuity, contrast sensitivity, pattern electroretinogram (ERG), and visual-evoked potentials, along with optic disc photography. While this study was too small to provide a definitive outcome, several of these test parameters indicated improved retinal function at the level of ganglion cells, consistent with inhibiting RGC apoptosis observed in the glaucomatous rats by NGF.
The pharmaceutical industry is considering other neurotrophic factors including ciliary neurotrophic factors (CNTF) for glaucoma rescue. Van Adel and colleagues have demonstrated that continuous administration of CNTF protects RGCs in animal models.3 Neurotech has initiated preclinical studies using its encapsulated cell technology (ECT) devices in which epithelial cells transfected with the CNTF gene are sequestered and release low but therapeutic levels for months’ to years’ duration. Efficacy of other neurotrophic factors for RGC rescue has been demonstrated,16 such as brain-derived neurotrophic factor (BDNF) which can protect RGCs following optic nerve crush in cat by intravitreal injection.17 BDNF also promotes regeneration of spinal cord axons following axotomy.18
Neuroprotection through the serotonin pathway
A promising area of neuroprotection research involves the serotonin (5HT) pathway. Memantine, in addition to its direct antiexcitotoxic effects as an NMDA receptor antagonist, is a noncompetitive antagonist at the 5HT3 receptor19 which may be part of the memantine neuroprotection mechanism for Alzheimer disease.20 Recent literature indicates that serotonergic compounds, such as the selective serotonin reuptake inhibitor antidepressant agents, potentiate neurite sprouting by NGF.21 The retina has 5HT receptors, but whether this putative mechanism has relevance to retinal and/or glaucomatous disease is not currently known.
Agonists that stimulate the 5HT system are neuroprotective for RGCs.22 Unfortunately, 5HT agonists have hallucinogenic action, and broad-action agonists at 5HT receptors would not be suitable for human glaucoma treatment. Among the interesting compounds in this regard is an indazole compound AL-34662 that is a selective 5HT receptor agonist at 5-HT-2A receptors and that lowers ocular pressure but which does not cross the blood–brain barrier.23
Neurotrophic factors for retinitis pigmentosa
Roy Steinberg and Matthew LaVail and their group24 made the seminal observation that neurotrophic factors could rescue photoreceptor cell death in an animal model of retinitis pigmentosa (RP). This observation in 1990 changed the face of research in photoreceptor neurodegenerative disease. Steinberg and LaVail and their colleagues showed that a single intraocular application of basic fibroblast growth factor (bFGF) delayed the loss of rod photoreceptors in the Royal College of Surgeons (RCS) rat. The rescue of the rods was panretinal, with a gradient of rescue highest near the injection site. This observation was subsequently extended to other genetic and environmental causes of photoreceptor cell death, including the light exposure damage model,25 in which the protection was shown to extend to other cytokines and neurotrophic factors beyond bFGF alone. These observations occurred during a time of great explosion of the field of photoreceptor degeneration research, including the laboratory creation of genetic models, including the P23H rhodopsin transgenic rat that mimicked human RP.26,27 Multiple neurotrophic factors have subsequently been found to rescue the rod cells in this rodent RP model.25
Exploration of this rescue proceeded along multiple lines, including cell biology of mechanisms,28,29 and by multiple delivery approaches, including application using gene delivery with adeno-associated virus (AAV) vector.30 Experiments with control animals subjected to mechanical injury by insertion of a “dry” needle into the retina31 also showed a surprising rescue of the rod photoreceptors. It was soon recognized that mechanical injury itself led to upregulation of expression of bFGF and CNTF. The precise mechanism of rescue was studied intensely but remained difficult to elucidate, and it is currently believed that the rescue is indirect and mediated through retinal glial cells which have receptors for neurotrophic factors, whereas the adult rods do not.28 Several of the factors activate the Jak-STAT pathway.28 Both NGF29 and CNTF30 were shown in other animal models to slow down photoreceptor loss quite significantly. NGF is the original neurotrophic factor identified in the 1950s by Rita Levi-Montalcini,32,33 who subsequently received the Nobel Prize for demonstrating that NGF acts on morphological differentiation of neuronal crest-derived cells and on peripheral and CNS neuronal cells. Cayouette and colleagues34 demonstrated that CNTF rescued the retinal degeneration slow (RDS) genetic mouse model of RP when applied by intraocular gene transfer that led to prolonged and stable CNTF expression in retina. They also showed that CNTF provided functional rescue of ERG potentials deriving from photoreceptor and bipolar cells and caused significant increase of the a-wave and b-wave of the rod-driven scotopic ERG.
CNTF protein and historical selection
CNTF was first identified by Ruben Adler et al. as a factor in chick embryo extract that could support survival of chick ciliary neurons.35 The factor was subsequently purified from the chick eye.36 CNTF is a small 200-amino-acid 22-kDa protein which assembles into an α-helical tertiary structure. Adler37 and McDonald et al.38 subsequently proposed that CNTF expression is upregulated as a stress factor and released from cells, although the mechanism is not understood as the protein lacks a secretion sequence.39
Proteins interact with cells through a cellular receptor. The CNTF receptor is a trimeric construct that includes the CNTF α-receptor, the leukemia inhibitory factor receptor-β (LIFR-β), and gp130 component.40 The CNTFRα soluble factor has been detected in cerebrospinal fluid and, consequently, a cell itself needs to express only LIFR-β and gp130. Subsequent binding of CNTF to this trimeric complex activates the Jak-STAT pathway41 leading to gene transcription.42 Ultimately, it is the upregulation of gene expression by CNTF that appears to be a crucial factor in human clinical trials of rod and cone rescue in photoreceptor degenerative diseases.
The effect of CNTF on nuclear activity was seen by AAV delivery of CNTF to RDS mice with peripherin mutation leading to slow death of rod photoreceptors.43 There were several seminal observations in this study, including a further demonstration that vector-mediated gene therapy provided long-term and panretinal rescue of rod photoreceptors following a single intraocular application. Second, this particular study showed a potentially adverse consequence of CNTF, as ERG response amplitudes were diminished in the treated eyes compared to the fellow uninjected eyes. Third, the rod cell nuclei showed an increase in euchromatin and an increase in the size, resulting in increased thickness of the outer nuclear layer (ONL), which is composed exclusively of photoreceptor cell bodies containing the nuclei. They concluded that CNTF upregulated gene expression in the photoreceptor cell either directly or, more likely, indirectly. This interpretation was quickly replicated in the rabbit retina by application of CNTF using a novel delivery mode of encapsulated cell-based intraocular devices (the Neurotech ECT)44 (Fig. 37.1). These devices release a continuous low level of CNTF protein into the vitreous, and hence, to the retina over weeks and months. Following implantation of ECT devices into the eye of normal rabbit, without retinal degeneration, the nuclear morphology changes, including dispersion of the nuclear chromatin, were consistent with DNA uncoiling that occurs during gene expression. The retinal function, as monitored by ERG recordings, was not affected, although high-dose CNTF suppressed the ERG for low-intensity stimuli. Cayouette et al. found that CNTF could augment retinal ERG function,34 and Bush et al. observed44 that CNTF did not adversely affect retinal function in lower dose, whereas the studies of Bok and colleagues43 note that high-dose CNTF expression with a vigorous chick β actin promoter suppressed retinal ERG function: these observations are consistant with CNTF having a safe biological range, beyond which it may cause potential retinal toxicity. A dose-ranging study in the rod–cone dystrophy 1 (RCD1) dog model of RP indicated that the ECT delivery approach spanned a sufficiently broad range of dosing that warranted safety study in a phase I human clinical trial for RP.
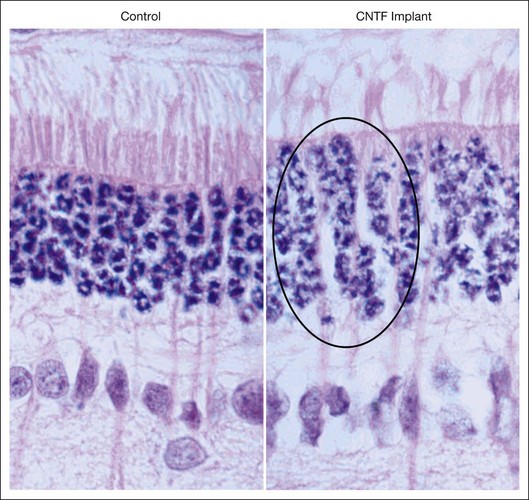
CNTF phase I trial for human photoreceptor degeneration
A human phase I trial of CNTF for RP neurodegenerative diseases was conducted and reported in 2006 using the Neurotech ECT device for intraocular production and delivery of CNTF protein.45 The device was surgically implanted in the eye of 10 study participants with advanced RP that severely limited their overall vision, including severe degradation of visual acuity (Fig. 37.2). The devices were implanted through a small pars plana incision which was closed by a single suture, causing minimal trauma to the eye, with the exception of one participant who had a transient choroidal effusion that resolved without ultimate reduction of baseline acuity. The devices were surgically explanted at the 6-month study conclusion. The initial 5 subjects received lower-dose implants delivering 0.28 ± 0.07 ng/day. The subsequent 5 participants received implants delivering 1.53 ± 0.54 ng/day, or about fivefold higher dose. The CNTF release levels were evaluated following explant. As this was a phase I safety study, the primary endpoints were safety parameters. No ocular or systemic complications ensued that met predetermined protocol adverse outcomes. The transfected cells derived from a human cultured retinal pigment epithelium (RPE) cell line. No serum antibodies to either CNTF or the transfected cells were detected in participants.
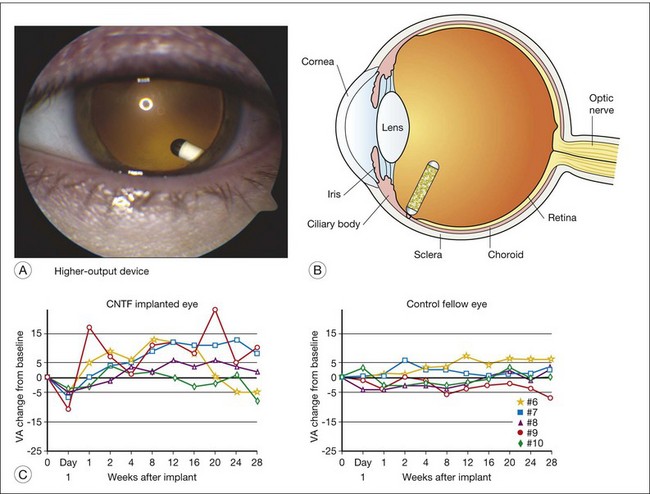
We considered the possibility of true biological rescue by CNTF and proposed a hypothesis of plausible action, as follows: visual acuity requires both close spacing of many photoreceptors and that these photoreceptors can functionally respond to light stimulation. We considered it unlikely that CNTF could “repopulate” the retina with new photoreceptors, but rather that CNTF might restore minimal function in cone photoreceptors that were present but were functionally dormant. Photoreceptors certainly can survive temporarily, even with no outer segments, in the rhodopsin knockout mouse model.46 These rod cells cannot respond to light in the absence of photopigment in the outer segments, and ultimately they die by approximately 90 days from birth. Second, both Bok et al.43 and Bush et al.44 generated evidence that CNTF elicited transcriptional activity in photoreceptor cells, consistent with CNTF stimulating higher metabolic output of photoreceptors. Finally, it is well understood that even relatively few cone photoreceptors can support some degree of visual discrimination, as deduced from retinal histology of postmortem eyes of individuals with RP and through psychophysical experiments that modeled photoreceptor loss.47 This chain of logic can be summarized as follows: first, photoreceptors become inactive before they die and go into a quiescent state and do not contribute to visual perception. Second, CNTF promotes photoreceptor transcriptional activity and metabolic function of cells, including elaboration of outer-segment material that supports photon capture and vision. Third, very few cone photoreceptors are required for a minimal level of visual acuity. Under these conditions, CNTF might partially restore visual function during a late stage of degeneration. This effect would be separate from CNTF preservation of photoreceptors in early stages of degeneration which was seen in at least 13 different animal models of degeneration, including phosphodiesterase (PDE) 6β mice, RDS peripherin mice, transgenic rats expressing P23H or S334ter mutant rhodopsin, RDY cat, RCD1 dog, rhodopsin knockout mice, and Q334ter rhodopsin transgenic mice. A study of CNTF in the S334ter rhodopsin transgenic rat model demonstrated regeneration of cone outer segments, consistent with the postulate of the CNTF phase I study group that CNTF may benefit vision by partially restoring or augmenting cone function.
Based on the successful safety study of CNTF protein released by encapsulated cell technology implants, phase II studies were developed to evaluate possible efficacy for age-related macular degeneration (AMD) involving geographic atrophy and for RP (David Birch, Retinal Foundation of the Southwest; Rafael Caruso, NEI/NIH; Paul Sieving, NEI/NIH; Weng Tao, NeuroTech; Santa Tumminia, NEI/NIH; Ronald Bush, NIDCD/NIH; and Richard Weleber, Casey Eye Instititue-OHSU). The scientific hypotheses were, first, whether one could demonstrate that CNTF slowed neurodegeneration and prolonged vision, and second, whether CNTF improves photoreceptor function in human RP. The CNTF-2 study for AMD trial had a principal outcome of visual acuity after 12 months’ treatment.48 The CNTF-3 and CNTF-4 were both for RP, with principal outcomes of visual acuity outcome at 12 months,49 or visual field sensitivity at 24 months,50 respectively. The trials involved about 60 subjects each, with asymmetric design, with 20 subjects receiving lower-dose implants and 40 subjects receiving higher-dose implants. All studies were fully enrolled.
The AMD trial (CNTF 2) reported results in 2011.51 No serious adverse events related to the CNTF implant or to surgical procedures occurred during the study. Biological activity of the implant was indicated by an increase of retinal thickness on optical coherence tomography for both the lower-dose and higher-dose CNTF groups, but not in the sham control eyes. Unfortunately, visual acuity outcome did not reach significance for the endpoint, but the higher-dose group showed a trend towards stabilization of best corrected visual acuity (BCVA) at 12 months compared to sham treatment (P = 0.078). Neither the ERG amplitudes nor Humphrey visual field sensitivity showed significant change, either for better or worse.49
New technology for endpoints for photoreceptor degenerations
In considering future trials involving disease that causes demise of rod or cone photoreceptors, the selection of clinical endpoints to demonstrate significant rescue has received intense consideration. Psychophysical measures of visual acuity, visual sensitivity, or extent of visual field appear to be insensitive measures, as they persist even after objective measures of ERG amplitudes are greatly reduced. A new potential endpoint involves adaptive optics scanning laser ophthalmoscopy, in which cone photoreceptors can be imaged in vivo by sharpening image clarity upon correction of optical aberrations inherent in the lens and cornea. Talcott et al.52 demonstrated the ability to image and count the cones in the foveal region of the human eye during CNTF treatment of 2 subjects. They found significant relative preservation of cone photoreceptors in the CNTF-treated eyes. While this approach currently is extremely labor-intensive, it may afford the best opportunity to track the natural history of photoreceptor loss by neuroprotective treatment trials in these slowly progressing diseases.
Delivery of neurotrophins
Neurotrophic factors are proteins of substantial size, and controlled delivery to target ocular tissues presents a formidable challenge. None of the neurotrophic factors are particularly amenable to crossing the blood–retinal barrier, nor can they be administered from outside the eye and achieve substantial molecular flow to intraocular tissue. While delivery of neuroprotective factors will also be considered in a separate section of this chapter, some mention is warranted here of the major approaches to delivery. Direct delivery of CNTF protein by intravitreal injection has worked well in rodent models. This approach has major limitations, however. The first is the limited half-life of therapeutic effect. A single intravitreal application in the rat eye of CNTF has a rapid clearance time. Despite this, the effect is longer-lasting than expected, following a single bolus injection, indicating that CNTF acts through a secondary mechanism, possibly by upregulating gene expression in glial cells which release some currently unknown protective factor. Persistence of treatment duration can be extended by repeated injections, as is done for anti-vascular endothelial growth factor (VEGF) compounds to treat neovascular AMD.53,54 However, even such heroic approaches are currently conducted only for 12–18 months, whereas neuroprotection for retinal photoreceptor degenerative diseases would need to continue for decades. Further, injecting a large bolus application of CNTF compound impacts retinal function and decreases the ERG amplitudes,43 whereas low-dose continuous CNTF release had the possibility of augmenting rod cell function.34
An alternative promising approach is ECT, in which low-dose protein is released continuously into the vitreous cavity following surgical implantation of the encapsulated cells transfected with the gene of choice, as used for the CNTF phase I clinical trial.45 Current indications from Neurotech are for continuous release in therapeutic quantity beyond 24 months duration. While surgical insertion of the device is a potential limitation, conversely, it is attractive to be able to remove the device by surgical explant if it is necessary to terminate the drug application.
Vector-mediated gene transfer by intravitreal or subretinal application also holds the potential for long-term chronic therapy. While adenovirus-mediated transfer has limited duration of expression of only many months, AAV vectors are capable of maintaining chronic release over the course of many years, as seen for the RPE65 trial in the dog.55 The current limitations of AAV vector-mediated transfer involve potential immunogenicity, potential for mutagenesis, and a current lack of regulation of gene expression level, including genetic control to terminate protein production. Quite likely, these limitations will be overcome in the future.
Antioxidants
Oxidative damage in light-induced and inherited photoreceptor degenerations
Absorption of light (the normal function of photoreceptor outer segments) increases oxidation of their lipids, creating morphological and functional damage as light exposure is increased.56–58 Both death and damage appear to be caused by oxidative stress, i.e., by the damaging effects of partially reduced forms of oxygen, often called reactive oxygen species. The idea that light-induced damage is caused by oxidative stress is supported by evidence that levels of endogenous antioxidants increase following light damage,58–60 and that exogenous antioxidants are protective60–67 for cones68,69 as well as rods.70–72 The stress-induced death of photoreceptors is accompanied by damage to the survivors.70,72
In several inherited photoreceptor degenerations the starting event is a mutation that leads to the death of rod photoreceptors. After the death of rods, the major consumers of oxygen in the retina, the tissue oxygen level in the outer retina is substantially increased,73 activating nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and inducing accumulation of superoxide radicals.74 Superoxide radicals attack macromolecules, causing oxidative damage and production of other molecules such as nitric oxide which generates peroxynitrite, a particularly damaging free radical that amplifies the damage.75 Over time the antioxidant defense systems and repair mechanisms in cones cannot compensate for damage, leading to severe cell damage and apoptosis. According to cone density distribution, cone dysfunction and death are expected to occur first in the periphery and then spread posteriorly, a process that is common to many inherited dystrophies. The generation of free radicals is relentless and, thus, any treatment directed at preserving cones must be provided continuously for the patient’s entire life. The repair mechanisms can protect from a limited amount of damage. The aim of antioxidant treatment is to reduce the rate of oxidative damage below the rate of repair in order to prevent dysfunction and apoptosis. To determine if this can be achieved, long-term experiments are required. Alternatively, evidence of protection could be provided in a shorter time length by the rescuing effect of a candidate drug on damaged but still viable cells (Figs 37.1 and 37.2).
Preclinical evidence of antioxidant protection in photoreceptor degenerations
Inhibitors of NADPH oxidase (Nox)
Usui et al.74 investigated the hypothesis that Nox and/or xanthine oxidase serve as critical intermediaries between increased tissue oxygen and the generation of excessive reactive oxygen species that cause oxidative damage to cones. Apocynin, a blocker of Nox, but not allopurinol, a blocker of xanthine oxidase, markedly reduced the superoxide radicals in the outer retina of the retinal degeneration-1 (rd1+/+) model of RP. Compared to rd1+/+ mice treated with vehicle, those treated with apocynin, but not those treated with allopurinol, had significantly less oxidative damage in the retina. Apocynin-treated, but not allopurinol-treated, rd1+/+ mice had preservation of cone cell density, increased mRNA levels for M- and S-cone opsin, and increased mean photopic b-wave amplitude. In Q344Ter mice, a model of dominant RP in which mutant rhodopsin is expressed, apocynin treatment preserved photopic ERG b-wave amplitude compared to vehicle-treated controls. These data indicated that Nox, but not xanthine oxidase, plays a critical role in the generation of oxidative stress leading to cone cell death in RP. Inhibition of Nox provides a new treatment strategy.
Nitric oxide synthase (NOS) inhibitors
Recent studies by Komeima et al.75 have shown that treatment of rd1 mice with a mixture of NOS inhibitors significantly increased cone survival, indicating that NO-derived peroxynitrite contributes to cone cell death. Treatment with 7-nitroindazole, a specific inhibitor of neuronal NOS, also significantly reduced cone cell death, but aminoguanidine, an inhibitor of inducible NOS, did not. These data suggest that NO generated by neuronal NOS exacerbates oxidative damage to cones in RP, and that combined therapy to reduce NO and oxidative stress should be considered.
Bolstering the endogenous antioxidant defense system
In another study by the same group,76 genetically engineered rd10 mice with either inducible expression of superoxide dismutase (SOD) 2, catalase, or both in photoreceptor mitochondria were generated. Littermates with the same genetic background that did not have increased expression of SOD2 nor catalase served as controls. Coexpression of SOD2 and catalase, but not either alone, significantly reduced oxidative damage in the retinas of postnatal day (P) 50 rd10 mice, as measured by protein carbonyl content. Cone density was significantly greater in P50 rd10 mice with coexpression of SOD2 and catalase together than rd10 mice that expressed SOD2 or catalase alone, or expressed neither. Coexpression of SOD2 and catalase in rd10 mice did not slow rod cell death. These data supported the idea of bolstering the endogenous antioxidant defense system as a gene-based treatment strategy for RP, and also indicated that coexpression of multiple components may be needed for effective neuroprotection.
Following the same line of research, Lu et al.77 explored the strategy of overexpressing components of the endogenous antioxidant defense system to combat oxidative damage in RPE cells and retina. In transfected cultured RPE cells with increased expression of SOD1 or SOD2, there was increased constitutive and stress-induced oxidative damage measured by the level of carbonyl adducts on proteins. In contrast, RPE cells with increased expression of glutathione peroxidase 1 (Gpx1) or Gpx4 did not show an increase in constitutive oxidative damage. An increase in Gpx4, and to a lesser extent in Gpx1, reduced oxidative stress-induced RPE cell damage. Coexpression of Gpx4 with SOD1 or 2 partially reversed the deleterious effects of the SODs. Transgenic mice with inducible expression of Gpx4 in photoreceptors were generated, and in three models of oxidative damage-induced retinal degeneration, increased expression of Gpx4 provided strong protection of retinal structure and function. These data suggest that gene therapy approaches to augment the activity of Gpx4 in the retina and RPE should be considered in patients with RP or AMD.
Carotenoids (lutein, zeaxanthin) in combination with other antioxidants
Miranda et al.78 have shown that the use of a combination of antioxidants delayed the degeneration process in rd1 mouse retina. In an effort to understand the mechanism of action of these substances (zeaxanthin, lutein, α-lipoic acid, glutathione, and Lycium barbarum extract) the changes in the levels of several proteins and oxidative stress markers in the rd1 retina were studied. The treatment increased glutathione peroxidase activity and glutathione levels and decreased cystine concentrations in rd1 retinas. Considering all the results obtained from treated and untreated animals, a high correlation was present between glutathione concentration and glutathione peroxidase activity, and there was a negative correlation between glutathione retinal concentration and the number of TUNEL-positive cells. No difference was observed between the numbers of nNOS and NADPH diaphorase-positive cells in treated and untreated rd1 mice. Thiol contents and thiol-dependent peroxide metabolism seem to be directly related to the survival of photoreceptors in rd1 mouse retina.
Rac1
Rac1 is a component of NADPH oxidase that produces reactive oxygen species.79 Rac1 is expressed abundantly in mammalian retinal photoreceptors, where it is activated in response to light stimuli.80 Knocking down Rac1 expression, by conditional gene targeting, in mouse rod photoreceptors resulted in protection against light-induced photoreceptor death compared with wild-type (WT) littermates.81 A similar protective effect on rods was also found by using apocynin, which inhibits NADPH oxidase activity. These results implicate both neuronal Rac1 and NADPH oxidase in cell death in this model of CNS degeneration. Diminished Rac1 expression in mouse rods had no effect on retinal structure or function examined by light microscopy, electron microscopy, rhodopsin measurement, ERG activity, and visual acuity, indicating that rod outer-segment morphogenesis proceeds normally in Rac1 conditional knockout mice. The lack of structural or functional effect of Rac1 depletion on photoreceptors, but protection under conditions of stress, indicates that the Rac1 pathway warrants exploration as a target for therapy in retinal neurodegenerative diseases.
Rod-derived cone viability factor
A new signaling molecule that represents a potential therapy for these currently untreatable diseases has been identified over the past 12 years.82 This protein, called rod-derived cone viability factor (RdCVF), maintains the function and consequently the viability of cone photoreceptor cells in the retina; mice that lack this factor exhibit a progressive loss of photoreceptor cells. The gene encoding RdCVF (Nucleoredoxin-like (Nxnl1) gene) also encodes, by differential splicing, a second product that has characteristics of a thioredoxin-like enzyme and protects both photoreceptor cells and, more specifically, its interacting protein partner, the tau protein, against oxidative damage. This signaling pathway potentially links environmental insults to an endogenous neuroprotective response. The Nxnl1 gene and, most likely, the paralogous gene Nxnl2 are part of a newly discovered redox signaling pathway that involves an enzymatic product and a trophic factor, both encoded by the same gene. It has been suggested82 that this therapy, by restoring a physiological signal, may efficiently treat the effects of a broad range of RP mutations. The delivery of RdCVF into the patient’s eyes can be achieved through different routes, by injection of the protein into the eye, expression from viral vectors, or delivery of RdCVF-producing cells that are encapsulated in a semipermeable membrane to avoid attack by the immune system. RdCVF is now in translation into a possible therapeutic agent to treat a spectrum of degenerative eye diseases.
N-acetylcysteine
Very recently, Lee et al.83 determined whether orally administered N-acetylcysteine (NAC) reduced cone cell death and preserved cone function by reducing oxidative damage in two models of RP, rd1 and rd10 mice. In rd10 mice, supplementation of drinking water with NAC promoted partial maintenance of cone structure and function for at least 6 months. Topical application of NAC to the cornea also reduced superoxide radicals in the retina and promoted survival and functioning of cones. Since oral and/or topical administration of NAC is feasible for long-term treatment in humans, and NAC has a good safety profile, it is reasonable to consider clinical trials to evaluate the effects of prolonged treatment with NAC in patients with RP.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


