Chapter 141 Molecular Genetics of Choroidal Melanoma
Introduction
While uveal melanoma represents only 5% of all melanomas, it is the most common primary intraocular malignancy of the adult eye, affecting approximately 5–11 individuals per million per year.1 Uveal melanomas arise from melanocytes within the uveal tract, which consists of the iris, ciliary body, and choroid of the eye. Iris melanomas are relatively benign; however, ciliary body and choroidal melanomas still present significant diagnostic and therapeutic challenges.2 Indeed, choroidal melanomas – the most common ocular melanoma – result not only in vision loss, but also in metastasis, which is uniformly fatal. Metastases most commonly target the liver, and the detection of hepatic metastatic lesions predicts a dismal outcome, with a median survival of only a few months.3 Unfortunately, despite advances in the diagnosis and treatment of the primary tumor, we have not witnessed a corresponding improvement in patient survival.
Current treatment for local disease, including eye-sparing approaches (e.g., radioactive plaque therapy, external beam radiation, laser therapy), often leads to profound loss of vision4; thus, the identification of novel targets could provide gene product-targeted therapeutic options, which may avoid local tissue destruction. To this end, it is essential that we determine the molecular mechanism(s) that promote the initiation and progression of uveal melanoma. In the past few years, emerging evidence suggests that a complex series of molecular steps occurs in which uveal melanocytes elude their anti-proliferative and pro-apoptotic harnesses to form a melanoma, and in up to half of patients, metastasize hematogenously to the liver and other organs.5 The high rate of metastasis in patients diagnosed with uveal melanoma despite local treatment is thought to be due to the presence of micrometastases that occur prior to diagnosis and treatment. These micrometastases can remain dormant, often for years or decades, before manifesting clinically as metastatic disease. It is therefore essential that we identify patients at risk for metastasis early, so that we may provide adjuvant systemic therapy in an effort to delay or perhaps prevent the progression to clinical metastatic disease. It is equally important that patients not at risk for metastasis are spared from unnecessary treatment with systemic chemotherapy with their attendant risks and side effects.
Cutaneous melanoma, uveal melanoma, and the ras/raf/mek pathway
Early work exploring the molecular genetics of uveal melanoma was based upon the genetic studies in cutaneous melanoma. Given their common melanocytic origin, observations made in cutaneous melanoma served to guide investigation into the molecular biology of uveal melanoma. Specifically, activating mutations in Ras and B-Raf had been shown to play a fundamental role in the development of cutaneous melanoma, occurring in more than 80% of these tumors.6 Mutations of Ras and B-Raf promote the activation of MEK1/ERK (or the mitogen-activated protein kinase (MAPK) pathway), thereby promoting cell proliferation and survival.7 Uncontrolled proliferation is a key feature of malignant transformation, and activation of the MAPK pathway is a common target for malignant progression. Early work demonstrated that the vast majority of primary uveal melanoma tissue exhibits immunohistochemical evidence of activation of the MAPK pathway.8,9 Based upon these promising findings, several groups have investigated the mutational status of Ras, B-Raf, and MEK1 in primary uveal melanomas, as well as in liver metastases from patients with uveal melanoma. Surprisingly, these studies have been overwhelmingly negative.10,11 Indeed, despite arising from the same cell type, there appear to be more dissimilarities between the molecular genetics of uveal melanoma and cutaneous melanoma than there are similarities.
GNAQ and GNA11 mutations in uveal melanoma
A recent breakthrough in our understanding of the development of uveal melanoma came from the discovery in uveal melanoma tissue of activating mutations in genes encoding for members of the GαQ family of large G proteins. Genetic screens have shown that approximately half of uveal melanomas exhibit mutations in the gene encoding for GαQ (GNAQ).12 Interestingly, over half of uveal melanomas lacking a mutation in GNAQ exhibit a mutation in the gene encoding for the related protein, Gα11 (GNA11).13 Of note, mutations in genes encoding other heterotrimeric G-protein alpha subunits have been reported in a variety of cancers.14 G proteins are a family of heterotrimeric proteins which signal from cell surface, 7-transmembrane spanning (G protein-coupled) receptors (GPCRs).15 GαQ and Gα11 have 90% sequence homology with each other but may have differing functional roles. Upon ligand binding to its GPCR, the GDP bound to the GαQ/11 subunit is exchanged for GTP, resulting in a conformational change and the subsequent dissociation of the GαQ/11 from the Gβγ subunits. These subunits are then able to regulate various second messengers. The GαQ/11 family mediates its activity through stimulation of phospholipase C-β (PLCβ), which leads to activation of protein kinase C (PKC), and ultimately activates downstream intracellular signaling pathways, including the MAPK signaling pathway. Thus, activating mutations in either GNAQ or GNA11 (GNAQ/11) may provide the link between uveal melanomas and MAPK activation.16
However, mutations in GNAQ/11 have not been found to correlate with clinical, pathologic, immunohistochemical, or genetic factors associated with advanced uveal melanoma.17 Mutations in GNAQ/11 have also been identified in blue nevi (benign intradermal melanocytic proliferations affecting the conjunctiva and periorbital skin).12 Although patients with blue nevi are at higher risk for uveal melanoma, blue nevi are not malignant. Moreover, activating mutations in GNAQ/11 have not been detected in cutaneous melanomas. Indeed, although activating mutations in GNAQ/11 can promote transformation of immortalized melanocytes, mutation of either gene is not sufficient for malignant transformation. Collectively, these findings suggest that mutations in GNAQ/11 may occur early during the development of uveal melanomas. Thus, although helpful in understanding early molecular events in the pathogenesis of this enigmatic tumor, GNAQ/11 is not likely to help identify patients who are at higher risk for later developing metastases. Nonetheless, as GPCRs are the target – directly or indirectly – of 50–60% of all present pharmacological agents,15 GαQ/11 may still provide a novel gene-product specific therapeutic target for the treatment of uveal melanoma.
Chromosomal abnormalities in uveal melanoma
Although several clinical and histological features of uveal melanoma have been associated with a poor prognosis, their ability to predict metastasis in an individual patient is limited. Conversely, specific chromosomal abnormalities appear to provide a more accurate predictor for metastasis and survival.18 Emerging evidence suggests that these specific chromosomal alterations occur either in metastasizing or non-metastasizing tumors relatively late in the progression of uveal melanoma. Several cytogenetic or chromosomal abnormalities (e.g., loss of 1p, loss of chromosome 3, gain of 6p, loss of 6q, loss of 8p, gain of 8q), have been linked statistically to metastatic death in uveal melanoma patients.19–22 However, among these markers, gain of chromosome 6p and loss of one copy of chromosome 3 (monosomy 3) have proved to be the most reliable predictors of metastasis and survival. Gain of chromosome 6p occurs mainly in nonmetastasizing tumors, and carries a better prognosis. Conversely, loss of one copy of chromosome 3 (monosomy 3) occurs most frequently in metastasizing tumors, and predicts a poor outcome.23 Of note, isodisomy 3, in which one copy of chromosome 3 is lost and the remaining (faulty copy) is duplicated, occurs in 5–10% of cases of patients with uveal melanoma and is prognostically equivalent to monosomy 3.24 Tumors with monosomy 3 have also been found to contain greater numbers of additional chromosomal abnormalities (aneuploidy) than tumors with disomy 3, suggesting that loss of chromosome 3 may promote the genomic instability observed in more aggressive tumors. This has complicated the interpretation of the prognostic significance of some chromosomal abnormalities in uveal melanoma. For example, although gain of copies of chromosome 8q has been reported in uveal melanoma and has been associated with a poor outcome, it may not be an independent risk factor for metastasis, but rather more common in tumors with monosomy 3, which is a very strong metastatic risk factor.25 Conversely, a late chromosomal alteration that does have independent prognostic significance is loss of chromosome 8p, which (in the setting of monosomy 3) predicts earlier metastasis and poorer survival.26
Gain of 6p, loss of 3: the genetic bifurcation in uveal melanoma
Interestingly, gain of chromosome 6p and monosomy 3 are almost completely mutually exclusive,21 representing a genetic bifurcation in uveal melanoma progression (Fig. 141.1). This observation has prompted efforts to use this unique molecular distinction to predict which patients are at higher risk for later developing metastases.
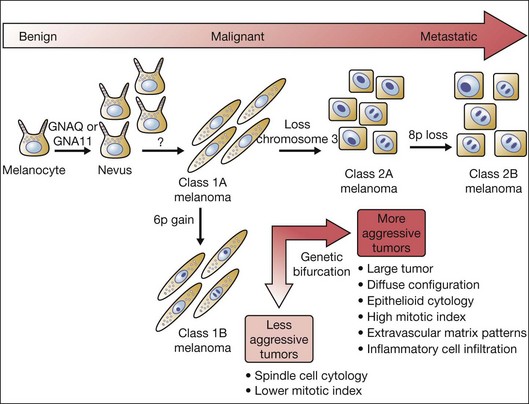
To distinguish between these two populations of uveal melanoma patients, initial efforts focused on cytogenetic analysis: standard karyotyping in which metaphase spreads are directly visualized and chromosomal abnormalities are identified by morphologic changes in chromosome size and banding pattern. However, this technique requires highly trained cytogeneticists, and the accuracy of the results is limited by sampling error attributable to analysis of only a few tumor cells, and to the inability to detect small genetic changes. Moreover, standard karyotyping is unable to identify isodisomy 3, resulting in a missed identification in up to 10% of uveal melanomas. Other techniques that also rely on direct analysis of intact chromosomes include fluorescence in situ hybridization (FISH), spectral karyotyping (SKY), and earlier forms of comparative genomic hybridization (CGH).27 Although these techniques provide some advantages over karyotyping, they are still prone to sampling error attributable to analysis of a small subset of tumor cells, and they are also unable to detect isodisomy.
More recent automated techniques (e.g., array-based CGH, microsatellite analysis (MSA), single-nucleotide polymorphism (SNP) analysis, multiplex ligation-dependent probe amplification (MLPA)), evaluate multiple regions across individual chromosomes using specific molecular probes. By allowing larger numbers of tumor cells to be examined, these techniques reduce the risk of sampling error, and increase the ability to detect smaller abnormalities compared with karyotyping and FISH. Additionally, MSA and SNP can detect the presence of isodisomy 3. However, the prognostic accuracy of these techniques compared with standard karyotyping is unclear.28
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


