Primary intraocular lymphoma (PIOL), also known as primary retinal lymphoma (PRL) or reticulum cell sarcoma, is a subset of primary CNS lymphoma (PCNSL) that involves the retina, vitreous, and optic nerve head with or without simultaneous CNS involvement. Most PIOLs are extranodal, non-Hodgkin, diffuse large B-cell lymphomas.
Epidemiology and Etiology
 More commonly seen in immunocompromised individuals
More commonly seen in immunocompromised individuals
 Incidence in the United States has tripled over the last 20 years both in immunocompromised and immunocompetent individuals.
Incidence in the United States has tripled over the last 20 years both in immunocompromised and immunocompetent individuals.
 100 new cases of PIOL each year
100 new cases of PIOL each year
 The incidence of PCNSL is 4 to 5 per 1000 person-years among patients with AIDS and 0.3 per 100,000 persons-years in immunocompetent patients.
The incidence of PCNSL is 4 to 5 per 1000 person-years among patients with AIDS and 0.3 per 100,000 persons-years in immunocompetent patients.
 25% of PCNSL patients have eye involvement at the time of presentation, whereas up to 85% of PIOL develop PCNSL.
25% of PCNSL patients have eye involvement at the time of presentation, whereas up to 85% of PIOL develop PCNSL.
 It typically affects an older population, usually in the fifth to sixth decades of life.
It typically affects an older population, usually in the fifth to sixth decades of life.
 Slight male preponderance
Slight male preponderance
 Bilateral in approximately 80%, but can be very asymmetric at presentation
Bilateral in approximately 80%, but can be very asymmetric at presentation
 Immunosuppression (secondary to AIDS or transplantation) is a risk factor. Infectious agents (Epstein-Barr virus, human herpesvirus 8, Toxoplasma) have been associated with PIOL. However, there have been no clear genetic or infectious markers to suggest susceptibility to PIOL.
Immunosuppression (secondary to AIDS or transplantation) is a risk factor. Infectious agents (Epstein-Barr virus, human herpesvirus 8, Toxoplasma) have been associated with PIOL. However, there have been no clear genetic or infectious markers to suggest susceptibility to PIOL.
Symptoms
 Blurry vision, floaters
Blurry vision, floaters
 Photophobia, ocular pain (rare)
Photophobia, ocular pain (rare)
Signs (Figs. 13-1 to 13-5)
 Vitreous cells (occurring in sheets) and haze
Vitreous cells (occurring in sheets) and haze
 Multifocal, cream-colored, subretinal infiltrates
Multifocal, cream-colored, subretinal infiltrates
 AC cells, keratic precipitates (KP)
AC cells, keratic precipitates (KP)
 Visual acuity is far better than expected based on the amount of inflammation
Visual acuity is far better than expected based on the amount of inflammation
Differential Diagnosis
 Sarcoidosis
Sarcoidosis
 Viral retinitis (cytomegalovirus, varicella zoster virus, herpes simplex virus), acute retinal necrosis
Viral retinitis (cytomegalovirus, varicella zoster virus, herpes simplex virus), acute retinal necrosis
 Toxoplasmosis
Toxoplasmosis
 Syphilis
Syphilis
 Tuberculosis
Tuberculosis
 Endophthalmitis
Endophthalmitis
 Metastatic cancer
Metastatic cancer
Diagnostic Evaluation
 The diagnosis is difficult, and requires malignant cells or tissue for diagnosis.
The diagnosis is difficult, and requires malignant cells or tissue for diagnosis.
 Fluorescein angiography demonstrates diffuse retinal pigment epithelium (RPE) perturbation and late staining at the RPE level with a granular or “mottled” pattern, early blockage with late staining of retinal/subretinal lesions, optic nerve staining or leakage, and pigment epithelial detachments. Lack of cystoid macular edema (CME) and retinal vascular leakage is an interesting feature of PIOL.
Fluorescein angiography demonstrates diffuse retinal pigment epithelium (RPE) perturbation and late staining at the RPE level with a granular or “mottled” pattern, early blockage with late staining of retinal/subretinal lesions, optic nerve staining or leakage, and pigment epithelial detachments. Lack of cystoid macular edema (CME) and retinal vascular leakage is an interesting feature of PIOL.
 Indocyanine green angiography shows small, round hypofluorescent areas that disappear in the late phase.
Indocyanine green angiography shows small, round hypofluorescent areas that disappear in the late phase.
 OCT: Nodular hyperreflective lesions in the RPE
OCT: Nodular hyperreflective lesions in the RPE
 Tissue diagnosis: Cytology (large atypical lymphoma cells), flow cytometry, cytokine analysis, and molecular analyses from ocular fluids (aqueous or vitreous)
Tissue diagnosis: Cytology (large atypical lymphoma cells), flow cytometry, cytokine analysis, and molecular analyses from ocular fluids (aqueous or vitreous)
 Brain/spinal MRI and lumbar puncture (LP), (for flow cytometry and cytology) to identify CNS involvement
Brain/spinal MRI and lumbar puncture (LP), (for flow cytometry and cytology) to identify CNS involvement
Treatment
 Current therapies are not curative.
Current therapies are not curative.
 Systemic chemotherapy is the mainstay of treatment even when associated PCNSL is not found. Adjunct local chemotherapy can be administered.
Systemic chemotherapy is the mainstay of treatment even when associated PCNSL is not found. Adjunct local chemotherapy can be administered.
 Systemic therapy: High-dose methotrexate; cyclophosphamide, adriamycin, vincristine, and prednisone; cytarabine; radiotherapy, and rituximab have all been tried with various degrees of success.
Systemic therapy: High-dose methotrexate; cyclophosphamide, adriamycin, vincristine, and prednisone; cytarabine; radiotherapy, and rituximab have all been tried with various degrees of success.
 Local therapy: Intravitreal methotrexate (400 mcg/0.1 mL), intravitreal rituximab (1 mg/0.1 mL), and radiation can all be considered.
Local therapy: Intravitreal methotrexate (400 mcg/0.1 mL), intravitreal rituximab (1 mg/0.1 mL), and radiation can all be considered.
 Recurrence and complications tend to be higher with radiation.
Recurrence and complications tend to be higher with radiation.
Prognosis
 Prognosis is poor.
Prognosis is poor.
 Median progression-free survival is less than 3 years, and overall survival is approximately 5 years and appears to be unaffected by treatment type.
Median progression-free survival is less than 3 years, and overall survival is approximately 5 years and appears to be unaffected by treatment type.
 Systemic spread outside of the CNS is extremely rare (<10% in autopsy specimens).
Systemic spread outside of the CNS is extremely rare (<10% in autopsy specimens).
 Ocular complications can include glaucoma, cataract (both due to inflammation or radiotherapy), retinal and/or optic nerve atrophy, vitreous hemorrhage, and retinal detachment.
Ocular complications can include glaucoma, cataract (both due to inflammation or radiotherapy), retinal and/or optic nerve atrophy, vitreous hemorrhage, and retinal detachment.
REFERENCES
Chan CC, Gonzalez JA. Primary Intraocular Lymphoma. Hackensack, NJ: World Scientific Publishing Co Pte Ltd; 2007.
Coupland SE, Damato B. Understanding intraocular lymphoma. Clin Exp. Ophthalmol. 2008; 36(6): 564–578.
Fardeau C, Lee CP, Merle-Béral H, et al. Retinal fluorescein, indocyanine green angiography, and optic coherence tomography in non-Hodgkin primary intraocular lymphoma. Am J Ophthalmol. 2009;147(5):886–894.
Grimm SA, McCannel CA, Omuro AMO, et al. Primary CNS lymphoma with intraocular involvement: International PCNSL Collaborative Group Report. Neurology. 2008;71:1355–1360.
Nussenblatt RB, Chan CC, Wilson WH, et al. International Central Nervous System and Ocular Lymphoma Workshop: recommendations for the future. Ocul Immunol Inflamm. 2006;14(3):139–144.
Sen HN, Bodaghi B, Hoang PL, et al. Primary intraocular lymphoma: diagnosis and differential diagnosis. Ocul Immunol Inflamm. 2009;17(3):133–141.
Figure 13-1. A. Diffuse, small- to medium-sized keratic precipitates in a patient with PIOL. B. There are large granulomatous-appearing pigmented KPs intermixed with stellate KPs more centrally.
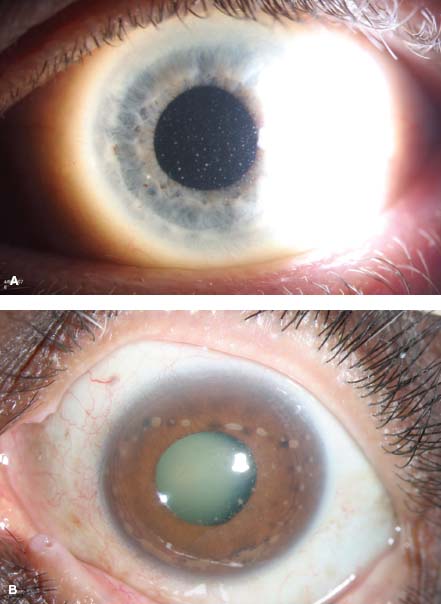
Figure 13-2. A 65-year-old white man developed PIOL and PCNSL. Vitreous cells in sheets are seen (inset).

Figure 13-3. Top: A 67-year-old Asian man with PCNSL and PIOL had large subretinal masses in the right eye and diffuse subretinal infiltrates in the left eye with significant vitreous haze. Bottom: The same patient following treatment with resolution of tumor infiltration.
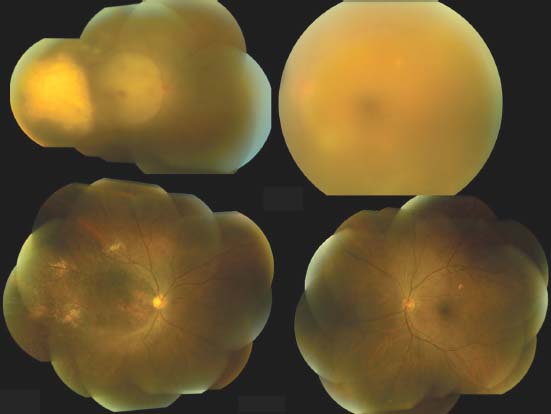
Figure 13-4. A, B. There is a large white-yellowish subretinal infiltrate temporal to the macula. The fluorescein angiogram shows granular (punctate) RPE perturbance throughout (C) and early blockage (D) . E. There is late staining of the large temporal infiltrate. F. MRI shows a solidly enhancing, apparently hypercellular mass in the left temporal lobe with surrounding edema. A brain biopsy was consistent with PCNSL.
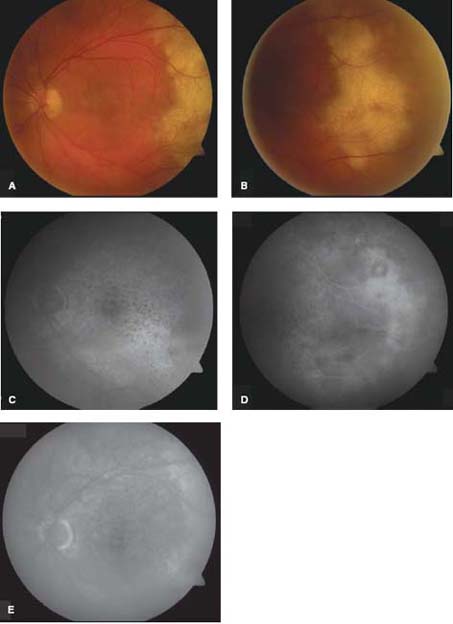
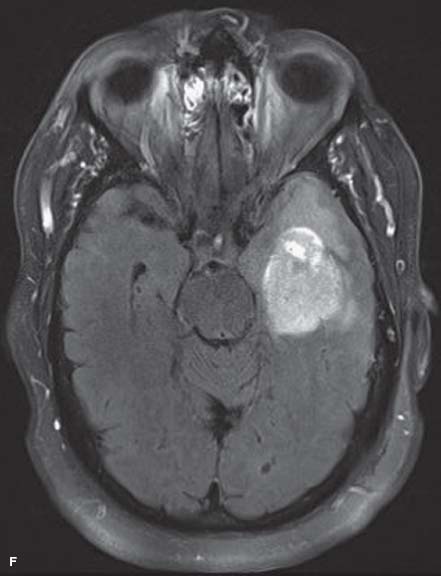
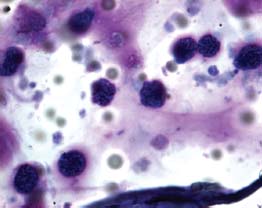
Figure 13-5. Vitreous cytology shows large lymphoma cells with scant cytoplasm and large convoluted nuclei.
RETINOBLASTOMA SIMULATING UVEITIS
Carol L. Shields
Retinoblastoma classically manifests as a solitary or multifocal, well-circumscribed retinal mass with dilated feeding vessels. Initially, this tumor appears as a subtle, translucent intraretinal mass. As the tumor enlarges, it assumes an exophytic, endophytic, or combined growth pattern. Exophytic retinoblastoma appears as a tumor with overlying subretinal fluid, whereas endophytic tumor appears as a mass with seeding into the vitreous cavity. A rare growth pattern is diffuse infiltrating retinoblastoma where the tumor assumes a relatively flat, ill-defined horizontal growth pattern along the retinal tissue, simulating uveitis or vitritis. Diffuse retinoblastoma seeds into the anterior segment with neoplastic pseudohypopyon and can cause intraocular hemorrhage, further confusing the findings. Often diffuse retinoblastoma is mistaken for uveitis.
Etiology and Epidemiology
 Retinoblastoma is the most common cancer of the eye in children. It is estimated that there are over 7000 cases per year worldwide with approximately 1800 in Africa, 4000 in Asia, 400 in Europe, 300 in the United States, and 600 in South America.
Retinoblastoma is the most common cancer of the eye in children. It is estimated that there are over 7000 cases per year worldwide with approximately 1800 in Africa, 4000 in Asia, 400 in Europe, 300 in the United States, and 600 in South America.
 The mortality rate from retinoblastoma varies depending on the continent, with 70% mortality in Africa, 40% in Asia, 20% in South America, and <>5% in Europe and the United States. Diffuse retinoblastoma represents <3% of all cases.
The mortality rate from retinoblastoma varies depending on the continent, with 70% mortality in Africa, 40% in Asia, 20% in South America, and <>5% in Europe and the United States. Diffuse retinoblastoma represents <3% of all cases.
 In the largest published report on this condition (34 cases), there were no cases of metastatic disease, as the eye was treated promptly.
In the largest published report on this condition (34 cases), there were no cases of metastatic disease, as the eye was treated promptly.
Symptoms
 Painless loss of vision
Painless loss of vision
 Floaters
Floaters
 Red eye with conjunctival injection
Red eye with conjunctival injection
 Often there are no complaints of vision loss, as young children tend to ignore visual compromise.
Often there are no complaints of vision loss, as young children tend to ignore visual compromise.
Signs (Figs. 13-6 and 13-7)
 Anterior segment
Anterior segment
 Iris neovascularization (50%)
Iris neovascularization (50%)
 Neoplastic pseudohypopyon (32%)
Neoplastic pseudohypopyon (32%)
 White tumor seeds on the corneal endothelium (24%)
White tumor seeds on the corneal endothelium (24%)
 Iris tumor nodules (18%)
Iris tumor nodules (18%)
 Cornea stromal edema (9%)
Cornea stromal edema (9%)
 Hyphema (9%)
Hyphema (9%)
 Posterior segment
Posterior segment
 Extensive ill-defined tumor infiltrating the retina (100%)
Extensive ill-defined tumor infiltrating the retina (100%)
 Average basal diameter of 20 mm
Average basal diameter of 20 mm
 Minimal retinal thickening (100%)
Minimal retinal thickening (100%)
 Extensive vitreous tumor seeds (91%)
Extensive vitreous tumor seeds (91%)
 Visible calcification (often subtle) on ultrasonography (79%)
Visible calcification (often subtle) on ultrasonography (79%)
 Visible calcification (often subtle) on CT (89%)
Visible calcification (often subtle) on CT (89%)
 Vitreous hemorrhage (24%)
Vitreous hemorrhage (24%)
Differential Diagnosis
 Endophthalmitis
Endophthalmitis
 Toxocariasis
Toxocariasis
 Toxoplasmosis
Toxoplasmosis
 Sarcoidosis
Sarcoidosis
 Juvenile idiopathic arthritis
Juvenile idiopathic arthritis
 Pars planitis
Pars planitis
Diagnostic Evaluation
 B-scan ultrasonography: Demonstrates retinal mass with possible intralesional dystrophic calcification. One must look closely for the calcification, as it might just be a fleck of calcium and not be obvious.
B-scan ultrasonography: Demonstrates retinal mass with possible intralesional dystrophic calcification. One must look closely for the calcification, as it might just be a fleck of calcium and not be obvious.
 Fluorescein angiography: Demonstrates dilated feeding artery and draining vein leading to a retinal mass. The mass might be minimally thickened and subtle.
Fluorescein angiography: Demonstrates dilated feeding artery and draining vein leading to a retinal mass. The mass might be minimally thickened and subtle.
 CT: Demonstrates a thickened intraocular mass, occasionally demonstrating intralesional calcification.
CT: Demonstrates a thickened intraocular mass, occasionally demonstrating intralesional calcification.
 MRI: Demonstrates the intraocular mass but will not show the calcification.
MRI: Demonstrates the intraocular mass but will not show the calcification.
 Fine-needle aspiration biopsy (FNAB): Should be reserved for cases that are diagnostically challenging. If retinoblastoma is present, tumor seeding into the orbit with risk for metastasis could occur as a result of the FNAB. This test should only be used if retinoblastoma is not high on the differential diagnosis. Usually FNAB is performed through the pars plana, but this should not be done in cases of possible retinoblastoma. Instead, the FNAB should be performed into the anterior chamber for an aqueous sample. Care should be taken not to seed tumor cells and cryotherapy should be performed at the entry site at completion of the procedure. If a vitreous specimen is needed, the pars plan route should be avoided. A preferred approach would be through the peripheral cornea, into the anterior chamber, through the peripheral iris, through the zonules (avoiding the lens), and into vitreous in an anteroposterior direction. Cryotherapy to the entry site is performed. This is a difficult procedure. Immediate cytologic preparation with preservative is important.
Fine-needle aspiration biopsy (FNAB): Should be reserved for cases that are diagnostically challenging. If retinoblastoma is present, tumor seeding into the orbit with risk for metastasis could occur as a result of the FNAB. This test should only be used if retinoblastoma is not high on the differential diagnosis. Usually FNAB is performed through the pars plana, but this should not be done in cases of possible retinoblastoma. Instead, the FNAB should be performed into the anterior chamber for an aqueous sample. Care should be taken not to seed tumor cells and cryotherapy should be performed at the entry site at completion of the procedure. If a vitreous specimen is needed, the pars plan route should be avoided. A preferred approach would be through the peripheral cornea, into the anterior chamber, through the peripheral iris, through the zonules (avoiding the lens), and into vitreous in an anteroposterior direction. Cryotherapy to the entry site is performed. This is a difficult procedure. Immediate cytologic preparation with preservative is important.
Treatment
 Most cases of diffuse retinoblastoma are extensive with tumor seeding into the vitreous cavity and anterior chamber, necessitating enucleation.
Most cases of diffuse retinoblastoma are extensive with tumor seeding into the vitreous cavity and anterior chamber, necessitating enucleation.
 Administration of intravenous or intra-arterial chemotherapy can be attempted, but the vitreous seeds might not completely respond and could show later recurrence.
Administration of intravenous or intra-arterial chemotherapy can be attempted, but the vitreous seeds might not completely respond and could show later recurrence.
 External beam radiotherapy can be employed, but a high recurrence of vitreous seeds is anticipated.
External beam radiotherapy can be employed, but a high recurrence of vitreous seeds is anticipated.
Prognosis
 Good if there is no invasion of the optic nerve, choroid, sclera, or orbit.
Good if there is no invasion of the optic nerve, choroid, sclera, or orbit.
 Poor if there is invasion of those structures. Adjuvant systemic chemotherapy to prevent metastasis should be given in cases of invasive retinoblastoma.
Poor if there is invasion of those structures. Adjuvant systemic chemotherapy to prevent metastasis should be given in cases of invasive retinoblastoma.
REFERENCES
Shields CL, Ghassemi F, Tuncer S, et al. Clinical spectrum of diffuse infiltrating retinoblastoma in 34 consecutive eyes. Ophthalmology. 2008;115:2253–2258.
Shields CL, Shields JA. Retinoblastoma management: Advances in enucleation, intravenous chemoreduction, and intra-arterial chemotherapy. Curr Opin Ophthalmol. March 2010;21:203–212.
Shields JA, Shields CL. Intraocular Tumors: An Atlas and Textbook. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 2008.
Figure 13-6. A young child with blurred vision and a red eye. A. A pseudohypopyon is noted. There are also subtle seeds along the pupillary margin at 9:00. B. Fundus evaluation shows diffuse vitreous tumor cells present. The vitreous cells are white and homogenous in appearance.
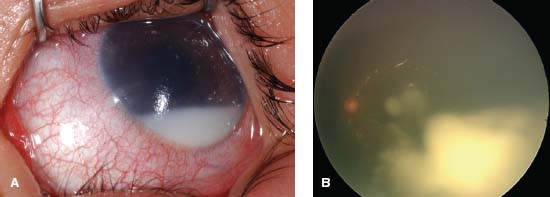
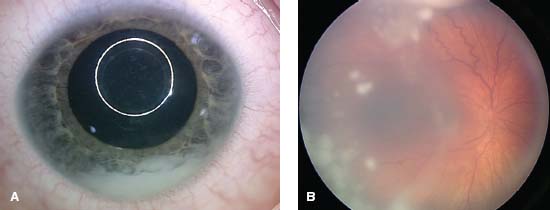
Figure 13-7. This child presented with a white eye and decreased vision. A. There is a white pseudohypopyon present. B. On fundus examination there are scattered vitreous seeds (opacities) that are stark white in color.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


