CHAPTER 125 Management of Parathyroid Disorders
The recorded history of hyperparathyroidism in modern medicine is relatively recent. Owen, a renowned British anatomist and curator, is generally acknowledged as being the first to describe the existence of the parathyroid glands in 1852.1 The discovery occurred subsequent to the death of the Zoological Society of London’s Indian rhinoceros, the postmortem of which was conducted by Owen and subsequently reported to the Zoological Society. In 1877, the Swedish histologist Sandstrom reported the existence of distinct glandular tissue adjacent to the thyroid in a dog.2 Over the subsequent 2 years, similar findings in other small mammals led to the search for, and ultimate discovery of, a similar organ in humans (glandulae parathyroideae), which Sandstrom reported on in 1880.
The earliest reports of clinical hyperparathyroidism involved bone disease, or osteitis fibrosa cystica, as termed by von Recklinghausen.3 These reports did not associate the characteristic changes in bone from hyperparathyroidism with parathyroid gland abnormalities, however. In 1903, Askanazy4 reported on an autopsy performed on a patient with osteomalacia and nonfusing long bone fractures in whom a large (>4 cm) tumor was seen adjacent to the thyroid gland, noting that it might represent a parathyroid tumor.
When Erdheim,5 a Viennese pathologist, discovered parathyroid gland morphologic and histologic abnormalities in patients with bone disease, an association between osteomalacia and parathyroid gland function was suspected. Erdheim studied the parathyroid glands by autopsy on all patients who died with bone disease, and noted that many patients with osteomalacia and osteofibrosis cystica showed enlarged parathyroid glands. He postulated that these glandular enlargements were secondary to compensatory hyperplasia, and that the bone disease was the primary initiating factor. Following up on initial experiments performed in rats by Gley, a French physiologist, Erdheim showed that cautery destruction of the parathyroid glands in rats produced not only tetany, as shown by Gley, but also the typical dental changes consistent with calcium not being laid down.5
Numerous reports of large parathyroid glands and bone disease followed until Schlagenhaufer6 suggested, at a meeting in Vienna in 1915, that if only a single parathyroid gland was thought to be enlarged, it should be excised. The event that followed this suggestion years later would usher in the future treatment of parathyroid disease. von Eiselberg,7 a pupil of Billroth, is noted as having performed the first parathyroid transplant. After performing total thyroidectomy in cats, von Eiselberg autografted the thyroid gland and a parathyroid gland into the animal’s abdominal wall. Postoperatively, the animals showed no sign of tetany, and, when subjected to histologic examination, these grafts showed evidence of neovascularization.7
Halsted’s experience with chronic hypocalcemia in thyroidectomy prompted him to study parathyroid transplantation experimentally in dogs.8 He showed that even very small portions of surviving parathyroid tissue autograft could be lifesaving in these animals, and tetany and death would result after removal. In addition, Halsted used intravenous calcium gluconate solution to treat animals after experimental thyroidectomy. These experiments and others sparked his ever-present mandate to perform thyroidectomy carefully and meticulously avoiding injury to the parathyroid glands and their blood supply. Halsted worked with Evans, a medical student at Johns Hopkins, to define the blood supply to the parathyroid glands using vascular casting technique, and emphasized that tetany after thyroidectomy was caused more by interruption of the vascular supply to the parathyroid glands than by their inadvertent removal.8
From a clinical standpoint, the treatment of parathyroid disease was to change significantly with the work of Mandle.9 Albert Gahne, a tram conductor, lived in Vienna after a bout with tuberculosis, which he acquired while serving in the Army during the years 1914-1918. He subsequently developed bone pain and muscle fatigue in 1921 from which he became disabled. In 1924, after a fall that resulted in a fractured femur, he came under the treatment of Mandle, who recognized these events as being consistent with parathyroid abnormality. Believing that this case might represent compensatory parathyroid hyperplasia as postulated by Erdheim, Mandle administered fresh parathyroid extract to Gahne without improvement. Subsequently, Mandle transplanted fresh parathyroid glands obtained from the victim of a street accident into Gahne, which again did not result in any resolution of symptoms. Recalling Schlagenhaufer’s suggestion of nearly 10 years earlier, Mandel explored the neck of the now severely crippled tram conductor and removed a parathyroid tumor.9 Gahne experienced a remarkable and immediate improvement in bone pain, and nearly 4 years later was walking with a cane free of pain. The disease recurred, and the patient was subsequently re-explored, but died after the procedure.10 This experience illustrated several important issues that would come to influence future work with surgical parathyroid disease, including clinical use of parathyroid transplantation in humans and the treatment of recurrent disease by re-exploration.
In 1924, Hanson developed a potent extract of the parathyroid glands, which when injected into animals led to increased serum calcium, decreased phosphate, and elevated output of calcium in the urine. When used chronically, this extract would produce osteoporosis in the animals.11
The association of elevated blood calcium levels and parathyroid dysfunction was well acknowledged when Charles Martell, a sea captain, was evaluated at the Massachusetts General Hospital in 1927, and found to have hypercalcemia and generalized demineralization of the skeleton believed to be caused by hyperparathyroidism. The first two of a total of six operations performed on Martell were done by Richardson, the Chief of Surgery at Massachusetts General Hospital. These first two neck explorations yielded only a single normal parathyroid gland on each side without identification of abnormal tissue.12 A third neck exploration was performed in New York in 1929 by Patterson without success. As renal function began to deteriorate with increasing symptoms of parathyroidism, Martell returned to Massachusetts General Hospital under the care of Albright and Cope. Cope had experience in several parathyroid explorations under the supervision of Churchill, and began cadaver dissection in preparation for re-exploration of Martell, which he did on three occasions in 1932 without success. At the urging of Martell, who had read extensively about his own disease and the potential locations of ectopic parathyroid tissue, Churchill planned a mediastinal exploration, the seventh surgical procedure on Martell. With Cope assisting, Churchill identified and removed most of a 3-cm tumor from the mediastinum, leaving an attached remnant portion with its vascular pedicle intact to avoid profound hypocalcemia. Despite these measures, tetany developed postoperatively, which required treatment with calcium supplementation. Several weeks after surgery, Martell experienced renal colic from an impacted ureteral stone, which required surgery. Martell died from laryngospasm after an operation to relieve obstruction from the impacted stone.
Although the series of procedures performed on Martell predated it and received more notoriety, the first successful parathyroidectomy performed in the United States was performed by Olsch in 1928 at Barnes Hospital of Washington University. In this instance, a large adenoma was removed precipitating a profound decline in serum calcium, which required massive doses of parathyroid extract and intravenous calcium to save the patient.12,13
Etiology and Pathogenesis of Hyperparathyroidism
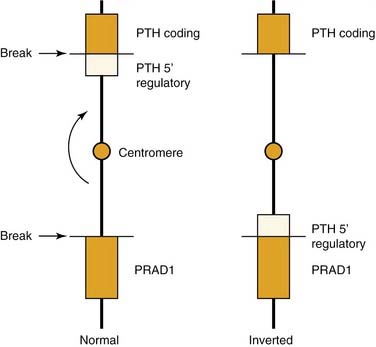
Parathyroid adenomas apparently are monoclonal or oligoclonal neoplasms, whereby the mechanism of propagation is believed to be clonal expansion of cells that have an altered sensitivity to calcium.14 The work of Arnold and colleagues14 indicates that the molecular events that seem to trigger clonal propagation are heterogeneous. The genetic mutational events that occur in hyperparathyroidism have been characterized in a few tumors. Among the events identified are genetic rearrangements of PRAD1, or parathyroid adenomatosis 1 oncogene, which is also known as cyclin D1. This proto-oncogene is located in the vicinity of the regulatory region of the gene for PTH production (Fig. 125-1).15,16 Subsequent realignment of DNA in this event now combines a growth promoter (PRAD1) with a regulatory region that would ordinarily control only PTH synthesis. This genetic realignment has not been uniformly shown in most parathyroid adenomas, with only a few having been shown to manifest rearrangement.
A more common molecular event, postulated to occur in parathyroid neoplasia, is alteration in tumor suppressor gene expression (Fig. 125-2). For this gene to be inactivated and product deficient, both alleles on the gene must be affected by the mutational event. Tumorigenesis occurs as a sequential event by inactivation of both copies of the suppressor gene.17 The most well known of these is the MEN1 tumor suppressor gene, which shows somatic mutations in both gene copies in 20% of patients with primary hyperparathyroidism.18 This gene was initially recognized in patients with multiple endocrine neoplasia type I (MEN-I) syndrome.19 Evidence of loss of suppressor gene function on chromosome 1p has been postulated as being an even more common event in the development of sporadic parathyroid adenomas. It has been suggested that patients with this chromosomal abnormality may be subject to developing the same constellation of endocrine changes found in MEN-I syndrome and perhaps at an earlier age.17 Suspected loss of tumor suppressor gene function has been identified in other chromosomal loci in patients with parathyroid adenomas including sites 15q, 9p, 6q, and 1q.17
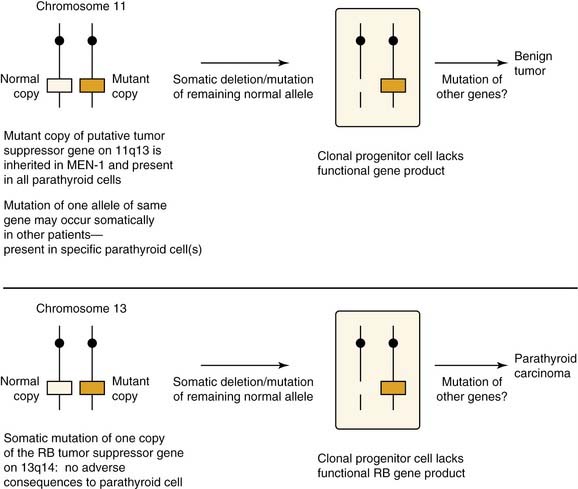
Figure 125-2. Hypothesized roles of inactivated tumor suppressor genes in the development of parathyroid neoplasia.
(From Arnold A. Genetic basis of endocrine disease 5: molecular genetics of parathyroid gland neoplasia. J Clin Endocrinol Metab. 1993;77:1108-1112. The Endocrine Society; reprinted with permission.)
Point mutations in the calcium-sensing receptor gene that reduced the activity of this gene have been elucidated as the basis for familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism.20 This gene seems to represent a likely candidate for molecular rearrangement and altered calcium sensing function in patients with hyperparathyroidism. To date, however, a calcium-sensing receptor gene mutation or allelic inactivation has not been shown in these patients. It has been postulated that alterations in the calcium-sensing function in primary hyperparathyroidism may represent postgenomic events related to reduced RNA transcript or the actual protein receptor in the parathyroid cell clone.21 Whether these events may represent the primary cause for, or secondary effect of, hypercalcemia remains to be determined. Abnormalities in the parathyroid cells vitamin D receptor similarly may represent changes occurring secondary to hypercalcemia and not a primary genetic event. An inactivating mutation in the gene coding for this receptor has been postulated in primary hyperparathyroidism, but results from these investigations are conflicting.22
Firm evidence supports the theory that ionizing radiation can represent the etiologic factor in several solid and humoral human cancers. Tisell and colleagues23 observed that there seems to be an association between exposure to ionizing radiation to the head and neck at an early age and the late development of hyperparathyroidism. This finding is supported by other independent observations where hyperparathyroidism has developed, presumably as a late complication of radiation therapy to the head and neck, similar to the finding noted with differentiated thyroid cancer.
Calcium Homeostasis and Parathyroid Hormone Secretion and Regulation
Calcium homeostasis is maintained by the complex interrelationship of PTH, vitamin D and its derivatives, and calcitonin. The polypeptide PTH contains 84 amino acids. When secreted by the parathyroid glands, PTH undergoes immediate degradation into the amino- and carboxyl-terminal fragments. The amino-terminal fragment is biologically active, but is rapidly cleared from the circulation, whereas the carboxyl-terminal fragment is biologically inert, is predominately cleared from the circulation by the kidney, and persists for a longer time particularly in patients with renal failure.24,25 The intact 1-84 molecule is the major circulating form of biologically active PTH; assays that measure the intact hormone more clearly reflect dynamic changes in PTH metabolism compared with the previous polyvalent assays, which measured a predominately inactive section of the PTH molecule.26
The major target end organs for PTH action are the kidneys, skeletal system, and intestine.27 PTH functions by binding to receptor sites in bone and kidney, which results in stimulation of the production of cyclic adenosine monophosphate (cAMP), which acts to carry out the cellular response of that specific target tissue to PTH.
The primary response to PTH by the kidney is to increase the tubular resorption of calcium and decrease the tubular resorption of phosphorus.28,29 The action of PTH on bone to regulate serum calcium is through the remodeling effect of osteoclast and osteoblast activity. The osteoblasts and their precursor cells in bone have a PTH receptor site, and binding to this site results in the production of cAMP. The osteoclasts do not have a PTH receptor site, but are stimulated indirectly through the cAMP response in the osteoblasts.30 The last important function of PTH occurs indirectly by increasing the rate of conversion in the kidney of 25-hydroxyvitamin D3 (calcifediol) to 1,25-dihydroxyvitamin D3 (calcitriol).31 The coordinated actions of PTH on bone, kidney, and intestine increase the flow of calcium into the extracellular fluid and as a consequence increase the serum calcium levels.
PTH is the primary regulator of rapid changes of extracellular calcium levels. The action of vitamin D affects delayed changes in calcium balance as opposed to the more immediate direct action of PTH.27 Calcitonin plays a much smaller role in calcium homeostasis. Calcitonin is secreted by the parafollicular cells of the thyroid gland and inhibits bone resorption. Calcitonin tends to be decreased in postmenopausal women, and may be increased by estrogen administration in these patients. Extremely high levels of calcitonin found in medullary carcinoma of the thyroid gland do not result in hypocalcemia.32
Several endogenous substances, including peptides, steroid hormones, and amines, have been found to influence PTH release.33,34 Calcium represents the most potent regulator of PTH secretion, however. Minor alterations within the physiologic calcium concentration range can induce considerable secretory responses—reduction of ionized plasma calcium by 0.04 mmol/L may elevate serum PTH by 100% or more. The rapid effect of extracellular calcium on PTH release suggests that calcium directly interferes with the release process, but the nature of this interference has been only partly clarified. It has been shown that external calcium mainly regulates secretion of newly synthesized hormone, which may bypass the few secretory granules in the parathyroid’s chief cells.35 Intracellular degradation with release of carboxy terminal PTH fragments occurs especially at high extracellular calcium concentrations; this attenuates the biologic activity of the secretory product because the calcium-regulating properties of PTH reside predominantly in its amino terminal portion.
Messenger RNA (mRNA) levels for PTH are increased within hours by low extracellular calcium, consistent with the effects of calcium on PTH secretion.36,37 Calcitriol (1,25-dihydroxyvitamin D3) reduces PTH mRNA levels and inhibits PTH secretion.38
Abnormalities in calcium-controlled PTH release represent a fundamental characteristic of the pathologic parathyroid tissue from hypercalcemic patients with parathyroid adenoma and hyperplasia of varying etiologies.39,40 The representative disturbance is characterized by variable calcium insensitivity of PTH secretion. The calcium insensitivity of ionized calcium and PTH release correlate to each other and to the degree of hypercalcemia in the patient.39
Intact PTH 1-84 is rapidly cleared from the human circulation and has a half-life of only a few minutes.41 Evidence does not exist to support dynamic regulation of peripheral PTH metabolism. PTH clearance mainly depends on high capacity uptake of Kupffer’s cells in the liver and on glomerular filtration. A small amount of PTH appears in the urine, however, because of tubular resorption and proteolysis. Circulating PTH is heterogeneous and contains various carboxy-terminal peptide sequences arising by cleavage, mainly at residues 33 to 43. Although approximately 15% of intact PTH 1-84 is metabolized to such circulating fragments, they make up at least half, and sometimes substantially more, of the immunoactive PTH in the circulation.42 The metabolism of such fragments depends virtually only on an intact renal function; consequently, they are accumulated to considerable degrees in renal insufficiency. In contrast, very few amino terminal PTH fragments exist in the circulation of euparathyroid individuals, although these fragments may become appreciable in primary and secondary hyperparathyroidism.
The diagnosis of primary hyperparathyroidism and evaluation of the extent or severity of secondary hyperparathyroidism have been facilitated by the development of immunometric “sandwich” assays. These assays use a pair of antibodies that recognize different regions of the PTH protein sequence.43–45 Because of cooperative effort of the antibodies, such a sandwich assay is more sensitive than either of the antibodies alone in displacement radioimmunoassays. With careful selection of antibodies, the immunometric assays are specific and sensitive for intact PTH, which allows identification of insufficient PTH secretion of hypoparathyroidism and a wide range of PTH levels without sample dilution. The immunometric analyzing process is swifter than the radioimmunoassays. With a reduction in the incubation times, the analysis may be accomplished in 15 to 30 minutes and, consequently, may be applicable intraoperatively.46,47
Clinical analysis with immunometric PTH assays usually discriminates hypercalcemic patients with hyperparathyroidism from patients with other causes of hypercalcemia. This discrimination is particularly evident with regard to malignancies of nonparathyroid origin, although approximately 5% to 10% of these patients show intact serum PTH in the low-normal range. Nonparathyroid tumors that produce intact PTH are exceptionally rare. These include ovarian and small cell carcinomas and thymoma.48–50
Parathyroid Anatomy and Histopathology
Normal Parathyroid Gland
The usual weight, size, and fat content of a normal parathyroid gland vary. The weight of a normal gland has been recorded to be as low as 40 mg, and a limit of 50 to 60 mg has been suggested.51 One study showed that the weights of normal parathyroid glands have a skewed distribution.51 The mean total weight in the study was 29.5 ± 17.8 mg, with an upper limit of 65 mg. The actual value for the 98th percentile was 75 mg, and this correlates with the operative findings typically noted in primary hyperparathyroidism.
The dimensions of normal glands are rarely mentioned in the literature. Normal dimensions of 3 to 6 mm in length, 2 to 4 mm in width, and 0.5 to 2 mm in thickness, and an average of three dimensions of 5 mm × 3 mm × 1 mm have been proposed.52 Normal glands measuring 12 mm and greater have been reported.53
The stromal fat content of parathyroid glands is the hallmark in the evaluation of their functional status. Detailed studies of normal glands have shown wide variations in fat content.54,55 The accepted percentage of normal fat content is approximately 50%. One study indicated that more than 75% of normal parathyroid glands had less than 30% stromal fat, 50% had less than 10%, and only a few had 40%.54 The variability of fat content reported by different studies suggests that measurement of stromal fat within parathyroid glands has become nearly useless as an indicator of function.54,55 In children and adolescents, parathyroid glands contain very sparse amounts of fat. After adolescence, stromal fat progressively increases until 25 to 30 years of age; subsequently fat content is largely determined by constitutional factors. Women seem to have a tendency to have higher glandular fat content, which may be related to total body fat concentrations.56
Four parathyroid glands is the usual number found in humans. In dissection studies of 428 human subjects by Gilmour,57 four parathyroid glands were found in 87% of all patients, and three glands were found in 6.3%. Akerstrom and coworkers58 reported comparable rates in an autopsy study of 503 individuals. In this study, four parathyroid glands were found in 84% of patients, and three parathyroid glands were found in 3%. The presence of supernumerary parathyroid glands is rare, and may have important clinical consequences, especially with respect to patients with hyperparathyroidism resulting from multiple-gland disease.
In a series of 2015 patients who were operated on for primary hyperparathyroidism, a hyperfunctioning supernumerary fifth parathyroid gland was the source of hypercalcemia in 15 patients (0.7%).59 Nine of these patients required reoperation to remove the supernumerary gland representing the parathyroid tumor. Most of these fifth gland tumors were located in the mediastinum, either in the thymus (seven tumors) or related to the aortic arch (three tumors). Edis and Levitt60 reported a rate of persistent hyperparathyroidism of 10% resulting from an enlarged supernumerary parathyroid gland in patients with secondary hyperparathyroidism. In a series of 762 patients with primary hyperparathyroidism, Wang and colleagues61 documented 6 patients with persistent hyperparathyroidism caused by hyperfunctional supernumerary glands (0.6%), all of which were located in or in close association with the thymus.
In the previously mentioned study by Gilmour,57 supernumerary parathyroid glands were observed in 29 of 428 specimens (6.7%). Five parathyroid glands were observed in 25 specimens (5.8%), 6 parathyroids were observed in 2 specimens (0.05%), 8 parathyroids were observed in 1 specimen, and 12 parathyroids were observed in 1 other specimen. From the studies by Gilmour57 and Akerstrom and coworkers,58 it is evident that supernumerary parathyroids are most commonly found within the thymus or in relation to the thyrothymic tract.
Parathyroid Gland Location
The location of parathyroid glands may vary, as a consequence of the variation in degree of migratory descent during development. Additional influences on these variable locations involves displacement of enlarged parathyroid glands during the development of hyperparathyroidism. Enlarged parathyroid glands tend to migrate in a fibroareolar plane, which offers little resistance as a result of gravity and the action of swallowing and variations in intrathoracic pressure.62
Eighty percent of the superior parathyroid glands are found at the cricothyroid junction approximately 1 cm cranial to the juxtaposition of the recurrent laryngeal nerve and the inferior thyroid artery.58 The superior parathyroids, which are intimately associated with the posterior capsule of the superior thyroid pole, are usually covered by an extension of the pretracheal fascia that envelopes the thyroid gland and connects it to the hypopharynx and esophagus and the carotid sheath. The relationship of these superior parathyroid glands with the pretracheal fascia is such that the glands themselves are allowed freedom of movement under this “pseudocapsule.” This feature discriminates parathyroid glands from thyroid nodules, which cannot move freely because they are enveloped by the true capsule of the thyroid gland.
Normal superior parathyroid glands may be found in the retroesophageal or paraesophageal space in approximately 1% of all instances.63 These spaces represent sites where enlarged superior parathyroid glands potentially descend to the superior and posterior mediastinum.
The incidence of intrathyroidal parathyroid glands is controversial. Akerstrom noted true superior intrathyroidal parathyroid glands in 3 instances (0.2%) among 503 autopsy specimens.58 Wang64 considered the superior parathyroid gland the most likely to be intrathyroidal primarily because of the close embryologic relationship of the primordium of the superior parathyroid gland with the lateral complex of the thyroid. In a series by Wheeler64a in which 8 intrathyroidal parathyroid tumors were noted in 7 of 200 patients (3.5%) undergoing neck exploration, 7 of these intrathyroidal glands were considered to originate from the inferior position. The overall incidence of intrathyroidal parathyroid glands ranges from approximately 0.5% to 3% as reported in the literature.65–68
Morphologic Characteristics of Parathyroid Glands
The visible discrimination between normal and abnormal hyperfunctioning parathyroid glands is essential to successful parathyroid surgery. The appearance of normal and abnormally functioning parathyroid glands varies and depends on the anatomic position and relationship to the thyroid capsule. Parathyroid glands that are located in loose connective tissue generally are more characteristically oval-shaped, bean-shaped, or teardrop-shaped. When parathyroid glands are closely juxtaposed to the thyroid capsule compressed by the pretracheal fascia, their appearance tends to be more conforming resulting in a flat shape with well-defined edges. The color of normal parathyroid glands ranges from yellowish brown to reddish brown. Generally, the color may depend on the amount of stromal fat, oxyphil cell concentration, and degree of vascularity.69 Normal glands tend to be more reddish brown or rust-colored in younger patients, whereas older individuals have parathyroid glands of a more yellow-brown or tobacco color. Enlarged hyperfunctional parathyroid glands have a color variation from dark brown to light yellow. Enlarged glands occurring in either secondary or tertiary hyperparathyroidism may have a lighter gray tone to the coloration. Parathyroid carcinoma can also show a mottled gray-to-white surface appearance.
Vascular Anatomy of the Parathyroid Glands
Normal parathyroid glands most commonly are supplied by a single dominant artery (80%).70 The length of the dominant artery supplying the parathyroid gland may vary from 1 to 40 mm. In most instances, the superior and inferior parathyroid glands derive their dominant arterial blood supply from the inferior thyroid artery. Ligation of the inferior thyroid artery during thyroid surgery may not always compromise the blood supply to the superior parathyroid gland, however. Abundant arterial anastomoses exist between the parathyroid glands and include anastomoses with thyroid arteries and dominant arteries of the larynx, pharynx, esophagus, and trachea. Of the superior parathyroid glands, 20% or more may be vascularized solely by the superior thyroid artery. In an autopsy study by Delattre and associates,70 10% of the inferior parathyroid glands derived their dominant arterial supply from a branch of the superior thyroid artery. In most of these instances, the inferior thyroid artery was noted to be absent. Primary mediastinal parathyroid glands have shown an arterial supply that represents a thymic branch of the internal mammary artery.71
Histopathology of the Parathyroid Glands
The parathyroid glands are enveloped in their own thin collagenous connective tissue capsule. This capsule extends septa into the gland, which separate the parenchyma into elongated chords or clusters of functional secretory cells. Blood vessels, lymphatics, and nerves travel along the septa to reach the interior of the gland.72
The major functional parenchymal cells of the parathyroid glands are the chief cells, which are slightly eosinophilic staining and measure 5 to 8 millimicrons in diameter. The chief cells contain many cytoplasmic granules (200 to 400 nm in diameter), which arise from the Golgi complex and represent the secretory granules.72 These granules contain PTH, which is synthesized from a precursor of prepro-PTH. With increasing age, the secretory cells of the parathyroid glands may be replaced by adipose cells, which may make up 50% to 60% of the gland in older individuals.
The second cell type making up parathyroid glandular parenchyma is the oxyphil cell. Although their function is unknown, it is believed that oxyphil cells and a third cell type, sometimes described as intermediate cells, may represent inactive phases of a single cell type.73 Oxyphil cells are less numerous, are larger (6 to 10 millimicrons in diameter), and stain more deeply with eosin than chief cells. Oxyphil cells tend to be more mitochondrial-rich, which may explain the increased ability of abnormal parathyroid glands with high oxyphil cell concentrations to concentrate technetium 99m (Tc 99m) sestamibi.74
By electron microscopy, chief cells show Golgi apparatus among dispersed granular endoplasmic reticulum and few secretory granules. The resting chief cells contain abundant lipid and glycogen, whereas during active phase the chief cells are smaller in size and contain decreased amounts of glycogen and lipid.75 The oxyphil cells are characterized by their large size and numerous cytoplasmic mitochondria.75
Single Glandular Enlargement or Parathyroid Adenoma
Single glandular enlargement, or adenoma, is the most common cause of hyperparathyroidism. Because of the variation in pathologic interpretation and patient population, however, the reported incidence of parathyroid adenoma ranges from 30% to 90%.76–80 In larger series of patients, where more uniformly accepted pathologic criteria were followed, approximately 80% to 85% of patients with primary hyperparathyroidism were found to have solitary parathyroid adenoma.76–78 Parathyroid adenoma may occur in any of the four parathyroid glands, but tends to involve inferior glands more commonly than superior glands.74
The gross appearance of parathyroid adenomas varies, but adenomas generally are oval-shaped or bean-shaped, red-brown in color, and soft in consistency.81,82 Adenomas may be bilobed or multilobulated in conformation. In 70% of adenomas, a rim of normal parathyroid tissue may be found around the hypercellular portion of the replaced normal gland. The absence of this characteristic does not exclude the presence of a parathyroid adenoma, however. The incised surface of an adenoma may appear smooth or nodular, or may show obvious areas of cystic change. Under light microscopy, adenomas appear similar to normal parathyroid glands, exhibiting a thin fibrous capsule with a cellular framework arranged in nests and cords invested by a rich capillary network. Other growth patterns include follicular, pseudopapillary, and acinar patterns.
Chief cells are the dominant cell types in most parathyroid adenomas. Oxyphil cells and transitional oxyphil cells are usually seen in varying proportions interspersed between the collections of chief cells.80–82 The chief cells in adenomas may be larger than found in normal glands, and may exhibit a greater degree of nuclear pleomorphism and giant cell formation.83,84 Nuclear atypia is of limited value, however, in distinguishing between parathyroid adenoma and carcinoma. Mitotic figures are uncommon in adenomas; however, they may be seen in a small percentage of cases.85
Variations in single glandular enlargement representative of parathyroid adenoma may occur and include the following subtypes: oncocytic adenoma, lipoadenoma, large clear cell adenoma, water clear cell adenoma, and atypical adenoma. Oncocytic adenoma is a rare subtype of parathyroid adenoma (4.4% to 8.4% of adenomas) that is composed predominantly (>80% to 90%) or exclusively of oxyphil cells.86,87 Previously, adenomas of the oncocytic variety were believed to be nonfunctional; however, oxyphil adenomas associated with hyperparathyroidism have been reported.88–90 Similar to typical adenomas, oncocytic tumors occur more frequently in women and are found most often in the sixth or seventh decade.89,90 Grossly, the tumors tend to be large, reported to range in size from 0.2 to 61 g; they are soft, spherical, ellipsoid, lobulated, or nodular, and range in color from light tan to dark orange-brown or mahogany.89,91 Microscopically, adenomas are composed predominantly of polygonal cells with abundant brightly eosinophilic granular cytoplasm and small round central hyperchromatic nuclei. Fat stain shows reduce cytoplasmic fat as per typical adenomas. Numerous mitochondria are densely packed throughout the cytoplasm on ultrastructural examination.
Lipoadenoma is another rare subtype of adenoma that was first described in 1958 as a parathyroid hamartoma.92 The initial description was that of a nonfunctioning mass; subsequent reports documented that these lesions can be responsible for hyperparathyroidism.93–95 The tumor comprises a lobulated yellow-tan mass composed of nests, acini, and cords of chief cells and occasional oxyphil and clear cells, intimately associated with large areas of adipose tissue or myxoid stroma or both. A rim of normal parathyroid tissue may be present at the periphery.
Water clear cell adenomas have been described, although their existence was initially doubted.96 In contrast to the large clear cell adenomas that accumulate glycogen, true water clear cell adenomas show a glycogen-free cytoplasm that is filled with membrane bound vesicles.97
Atypical adenoma is the term used to describe parathyroid adenomas that exhibit atypical cytologic features without definite evidence of malignancy (i.e., vascular or soft tissue invasion or both or metastases).98 It is important to distinguish these benign lesions from parathyroid carcinoma. The malignant potential of atypical adenomas in terms of recurrent or metastatic behavior is uncertain. These lesions may exhibit conspicuous mitoses, adherence to surrounding tissues, trabecular cellular arrangements, capsular invasion, or broad fibrous bands.99 One such lesion described consisted of a multifocal spindle cell proliferation that was mitotically active, averaging eight mitoses per 10 high-power fields within an otherwise typical parathyroid adenoma.100
Multiple Enlarged Glands or Parathyroid Gland Hyperplasia
Primary parathyroid hyperplasia is defined as proliferation of the parenchymal cells leading to increase in gland weight in multiple parathyroid glands in the absence of a known stimulus for PTH secretion. Two types of parathyroid hyperplasia are seen: the common chief cell hyperplasia and the rare water cell or clear cell hyperplasia.76,77
Chief Cell Hyperplasia
In 1958, Cope and associates101 first showed chief cell hyperplasia as a cause of primary hyperparathyroidism. It accounts for approximately 15% of hyperparathyroidism in reported series; however, some reports have indicated that about half of primary hyperparathyroidism may be produced by hyperplasia. A variation in this reporting is generally attributable to discrepancies in pathologic interpretation of abnormal parathyroid glands. The stimulus for this disorder is unknown; some studies have indicated a role of a possible circulating factor that can induce proliferation of parathyroid cells in culture. Approximately 30% of patients with chief cell hyperplasia have some type of familial hyperparathyroidism or one of the MEN syndromes.76,77,102,103 Molecular studies have shown that hyperplasias are monoclonal proliferations.15,16
Grossly, there is enlargement of all four glands. The glands may be of variable size, or they may be uniformly enlarged. By light microscopy, the dominant cell types are chief cells; however, one may also observe intermixed oxyphil cells and transitional oxyphil cells. The cellular proliferations may also give rise to nodular formation, and this can cause asymmetric gland enlargement.76,77
The amount of cytoplasmic fat in the chief cells is either reduced or absent.76,77 The chief cell in the nodular areas may be totally devoid of any fat, whereas the cells between the nodules may contain fat. Abnormal nuclei or mitoses are distinctly rare.77
Water Clear Cell Hyperplasia
Water clear cell hyperplasia is rare and is characterized by a proliferation of vacuolated water clear cells in multiple parathyroid glands. It shows a female predilection and leads to pronounced hypercalcemia and severe clinical disease. This represents the only parathyroid disorder in which the superior glands are larger than the inferior parathyroid glands. The glands affected by water clear cell hyperplasia tend to be larger and more irregular in shape with lobular extensions to surrounding soft tissue. By light microscopy, the glands show diffuse proliferations of clear cells characterized by clear cytoplasm and small dense nuclei. On high-power magnification, the cytoplasm is shown to be filled with small vacuoles. Cytoplasmic lipid is generally not present; however, moderate amounts of glycogen may be identified. The histologic appearance of water clear cell hyperplasia bears a resemblance to that of renal cell carcinoma.104
Secondary parathyroid hyperplasia as a consequence of renal failure cannot be distinguished from primary hyperplasia with the exception that early in the disorder there seems to be a greater tendency for the glands to be more uniform in size.105 As the disease progresses, asymmetry becomes more evident in renal failure–induced disease. The degree of glandular enlargement tends to reflect the severity of the underlying renal disorder.106 The largest glands are noted in patients whose renal disease began in childhood.107
Parathyroid Carcinoma
Parathyroid carcinoma is a rare malignant neoplasm derived from the parenchymal cells of the parathyroid glands. It has been reported to be responsible for 0.1% to 5% of cases of primary hyperparathyroidism.108–112 It is uncertain whether parathyroid carcinoma actually begins within preexisting benign parathyroid lesions.112,113 Carcinoma has been postulated to arise in the setting of primary parathyroid hyperplasia, notably familial hyperplasia.114–116 Only rare patients with parathyroid carcinoma have a history of prior neck irradiation.110,112
Morphologic features diagnostic of parathyroid malignancy are difficult in terms of definition and practical application during surgery. In one series of 40 patients with metastatic parathyroid cancer, 50% were thought to have benign disease by the operating surgeon and consulting pathologist during the time of initial exploration.117 Metastases are the only certain sign of malignancy. Metastatic behavior at the time of presentation is distinctly rare, however.108
Parathyroid carcinomas are characteristically large tumors, with 30% to 50% being palpable at the time of presentation.108,109 The tumors may measure 6 cm in diameter with a mean of approximately 3 cm.108 Although the average weights of carcinomas are reported to be greater than those of adenomas, there seems to be great overlap, indicating that weight alone may not be a major distinguishing characteristic between benign and malignant lesions. Carcinomas generally arise in the usual parathyroid locations, although they have been described in ectopic supernumerary glands within the mediastinum.118 Most parathyroid carcinomas are firm or hard in consistency and have a gray-to-white surface color as opposed to adenomas, which tend to be soft and tan in appearance. Adherence of the lesion to surrounding tissues is common, and these glands may be noted to extend to involve the soft tissues around the thyroid gland or the thyroid parenchyma itself. This may not prove to be a valuable differentiating feature because previous hemorrhage into a benign adenoma may be associated with fibrosis and adherence to adjacent structures in benign disease.112
Metastases at the time of presentation are unusual, but may rarely be found in regional lymph node basins.117 As opposed to regional metastases, parathyroid carcinoma is more often associated with widespread local infiltration, with invasion into contiguous structures, such as thyroid gland, strap muscles, trachea, and recurrent laryngeal nerve. Advanced metastases may occur and may be found in the lungs, bone, cervical and mediastinal lymph nodes, liver, occasionally kidney, and adrenal glands.117,119 Pulmonary metastases are the most common distant metastasis noted.117
Microscopic diagnosis of parathyroid carcinoma is a difficult task. The entire gland is traversed by broad fibrous bands that seem to originate from the capsule and extend into the substance of tumor leading to a lobulated appearance. The cells may be clear or rarely oxyphilic, and are arranged in nests and trabeculae.120 The cell may be uniformly bland or may show metaplasia, but the cases with minimal atypical findings may be difficult to distinguish from an adenoma.121–123
Mitosis can be seen in most instances and has been suggested as a primary factor in diagnosing parathyroid carcinoma.124 Mitotic figures may also be seen in parathyroid adenoma and hyperplasia, however, and their absence does not rule out a diagnosis of carcinoma.125,126 It is generally acknowledged that mitoses in parathyroid lesions should be of concern, especially because the follow-up in reported benign cases of parathyroid tumor exhibiting mitoses has been limited. Increased mitotic activity in unequivocal parathyroid carcinoma is an indicator of poor prognosis.127
The only reliable indicators of malignancy in parathyroid carcinoma are invasion of the surrounding structures, metastases, or both.127 Features such as desmoplastic reaction, mitotic activity, nuclear atypia, and necrosis may be more common in carcinoma than in benign lesions, but do not constitute criteria sufficient for a diagnosis of malignancy.127 In the absence of an infiltrative growth pattern, the parathyroid lesion showing some other feature of malignancy including mitoses may be designated as atypical adenoma. 128 Nonfunctioning parathyroid carcinomas have been rarely described, tending to be large and consisting of clear or oxyphil cells.129,130
Parathyroid carcinoma usually grows slowly and can be an indolent tumor. Multiple recurrences after surgical resection are common and may occur over a 15- to 20-year period.121,122 Patients with parathyroid carcinoma often die as a result of the effects of excessive PTH secretion and uncontrolled hypercalcemia, rather than growth of the tumor mass. Surgical excision of recurrence or metastases may provide excellent palliation by reducing tumor burden and consequently hormone production.131,132
Surgical Embryo-Anatomic Relationships in the Central Neck
Parathyroid tissue originates from primordial pharyngeal endoderm formed in the third and fourth pharyngeal pouches during the fifth week of embryologic development (Fig. 125-3). The epithelial lining of the dorsal wing of the third pharyngeal pouch differentiates into primordial parathyroid glandular tissue, whereas the ventral portion of the pouch differentiates into the thymus. As the thymus migrates medially and inferiorly, it pulls the inferior parathyroid gland (parathymus) with it into the thymic tail. Eventually, the main portion of the thymus migrates to its final position in the upper thoracic region, and its tail involutes, leaving the developing parathyroid gland to come to its position on the dorsal surface of the inferior pole of the thyroid gland. This glandular tissue eventually forms the inferior parathyroid gland. Simultaneously, the epithelium of the dorsal wing of the fourth pharyngeal pouch begins to differentiate into parathyroid glandular tissue. After separation from the regressing pouch, it becomes associated with the lateral portion of the caudally migrating thyroid, and is carried a short distance medially and inferiorly until it resides posterior to the superior pole of the thyroid gland. This tissue eventually develops into the superior parathyroid gland.
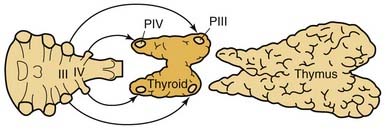
Figure 125-3. Embryologic derivation and subsequent descent of the parathyroid glands with associated structures.
This embryologic pattern of development has significant implications for the identification of ectopic or normal glandular variance during the course of parathyroidectomy. The longer embryologic migration results in an extensive area of potential dispersal for the normal inferior parathyroid gland. In 61% of cases, the glands are situated at the level of the inferior poles of the thyroid lobes on the posterior, lateral, or anterior aspects. In 26% of cases, they are situated in the thyrothymic ligament or on the upper, cervical portion of the thymus. More rarely, in 7% of cases, they are situated higher up, at the level of the middle third of the posterior aspect of the thyroid lobes, and may then be confused with the superior parathyroid gland. Because the embryonic descent of the thymus extends from the angle of the mandible to the pericardium, anomalies of migration of the parathymus, whether excessive or insufficient, are responsible for high or low ectopic locations of the inferior parathyroid gland. The incidence of higher ectopia along the carotid sheath, from the angle of the mandible to the lower pole of the thyroid, does not seem to exceed 1% to 2%.133–135 Alternatively, if the separation from the thymus is delayed, the inferior parathyroid gland may be pulled inferiorly into the anterior mediastinum to a varying degree. In this circumstance, they are usually within the thymus, at the posterior aspect of its capsule, or still in contact with the great vessels of the mediastinum. Lower ectopic regions such as these are noted in 3.9% to 5% of instances.134,136
The superior parathyroid glands follow the thyroid migration of the ultimobranchial bodies, which travel toward the lateral part of the main medial thyroid rudiment. In contradistinction to the inferior glands, the superior parathyroids have a limited descent within the neck. They remain in contact with the posterior part of the middle third of the thyroid lobes. This limited course of embryonic migration explains why they remain stable in their regional distribution when not pathologic. In most instances, they are grouped at the posterior aspect of the thyroid lobes, in an area 2 cm in diameter, whose center is situated about 1 cm above the crossing of the inferior thyroid artery and the recurrent laryngeal nerve.134,137 Because of the more extensive descent of the inferior parathyroid gland, the descent of the parathymus result in the inferior and superior glands crossing during development. This embryonic crossing of the glands explains why their grouping at the level of the inferior thyroid artery, at the junction of the middle and inferior thirds of thyroid lobe, is in many respects quite close, depending on the degree of migration of the inferior parathyroid gland.
Because of the short migratory descent of the superior parathyroid gland, the area of dispersal of these glands is limited, and congenital ectopic positions of the superior gland are unusual. In 13% of instances, the glands are on the posterior aspect of the superior pole of the thyroid lobe in a laterocricoid, lateropharyngeal, or intercricothyroid position, and, exceptionally, in less than 1% of instances, they are above the upper pole of the lobe. In 1% to 4% of instances, they are clearly posteriorly based behind the pharynx or esophagus. Parathyroid glands that are found in the posterior superior mediastinum are usually neoplastic superior parathyroid glands that have migrated as a consequence of gravity and changes in intrathoracic pressure.137
Clinical Features of Primary Hyperparathyroidism
Incidence
The incidence of hyperparathyroidism has changed dramatically over the past 3 decades. In the early 1970s, before the widespread use of multichannel autoanalysis of blood chemistry, Heath and colleagues138 reported an annual incidence of 7.8 cases/100,000 in Rochester, Minnesota. After the introduction of routine serum calcium assessment later in the 1970s, the incidence rate increased to 51 cases/100,000/year. After the most prevalent or clinically significant cases were managed, the incidence declined to 27 cases/100,000 annually, and more recent reports indicate that within the population associated with this area of Minnesota, a steady decline in the incidence of hyperparathyroidism has been noted since the late 1970s. This decline cannot be explained by the more limited routine use of multichannel autoanalyzing of blood, which has become part of cost-saving measures since the late 1990s; also, this declining incidence has not been similarly experienced in other populations nationally or internationally.
A higher rate of incidence was noted in the Stockholm study, which examined more than 15,000 subjects over a 2-year period (1971-1973) with a follow-up at 10 years. The early rate was assessed at 6 cases/1000, which at the 10-year follow-up was verified to be 4.4 cases/1000. This rate has not changed appreciably over a 20-year period, in contrast to the Rochester experience.139 One may postulate that the Rochester experience is unique in that after obtaining a very high incidence rate promulgated by routine calcium screening, higher prevalence cases were eliminated to reveal a lower base incidence rate in a population that receives its treatment at a single major center, explaining the steady decline in this fixed population base.
There seems to be a distinct predilection for a higher incidence of hyperparathyroidism in women, especially women beyond menopause. The highest prevalence rate among women in the Stockholm experience confirmed at the 10-year follow-up was approximately 13/1000, which represented a female-to-male ratio of about 4 : 1. This experience is similar to that of other published reports.138,140–143 Several of these studies based on serum calcium screening have shown that the prevalence of primary hyperparathyroidism in the general population is 0.1% to 0.3%, and in women older than 60 years, the prevalence is more than 1%.138,139,144 The clear female preponderance points to the fact that every woman has a 1% risk of experiencing primary hyperparathyroidism during her lifetime. It has been estimated, however, that about 90% of individuals with primary hyperparathyroidism remain undiagnosed.145 The screening of serum calcium has been a particularly important factor leading to the detection of patients with mild symptoms or no symptoms, especially among postmenopausal women.
Presentation
The concept of clinical features associated with primary hyperparathyroidism has changed during recent years. So-called classic, specific symptoms (i.e., bone disease, renal stones, and hypercalcemic crisis) represent obvious manifestations of the disease. The relative proportion of patients with osteitis fibrosis cystica, nephrolithiasis, and hypercalcemic crisis has continuously decreased in clinical series because of the increase in the number of patients with nonspecific or nonapparent symptoms. Currently, osteitis fibrosis occurs in about 1% of patients, and only 10% to 20% of patients have renal stones.146–148 Nonspecific symptoms include malaise, fatigue, depression and other psychiatric symptoms, sleep disturbance, weight loss, abdominal pains, constipation, vague musculoskeletal pains in the extremities, and muscular weakness.
The term asymptomatic hyperparathyroidism has been commonly applied when the disorder is detected during health screenings and population studies, or coincidentally during medical examinations. The usual presenting abnormality in these patients is an abnormally elevated serum calcium detected on routine blood chemistry screening. Despite the lack of obvious abnormalities noted at the time of diagnosis, caution should be exercised before declaring that a patient is asymptomatic. Many seemingly asymptomatic patients may manifest subtle or even “silent” sequelae of hyperparathyroidism at the time of presentation, and these patients may be more appropriately described as minimally symptomatic because nonspecific symptoms and unidentified complications of hyperparathyroidism would be eliminated by parathyroidectomy.149 Some of these nonspecific sequelae include emotional complaints, muscular fatigue, constipation, bone and joint pain, and silent objective findings such as asymptomatic renal calculi and decreased bone mineral density. In most patients with asymptomatic or minimally symptomatic hyperparathyroidism, these symptoms are subtle and may be so common in the general population that they preclude establishment of a causal relationship to primary hyperparathyroidism.
Kidney and Urinary Tract
Historically, greater than 50% of patients with hyperparathyroidism develop renal symptoms manifested by nephrolithiasis and nephrocalcinosis. This percentage decreased significantly to approximately 4% after the widespread use of screening tests for serum calcium levels.150 Most stones are composed of calcium oxalate; however, calcium phosphate stones may also occur. The symptoms associated with urolithiasis include renal colic, hematuria, and pyuria. Metabolic acidosis may also be a part of the clinical syndrome.
Skeletal System
Bone loss in hyperparathyroidism occurs at cortical bone sites generally sparing trabecular bone.151 Because of this finding, the role of hyperparathyroidism in osteoporosis is unclear, especially for patients in whom symptoms are minimal or absent, and in whom the disease is mild. Postmenopausal women with primary hyperparathyroidism exhibiting early signs of osteoporosis seem to be at significant risk for developing more severe bone disease and resultant sequelae (i.e., vertebral and hip fractures). The benefit of parathyroidectomy is most apparent in these patients.152
Neuromuscular System
Muscle weakness, particularly in the proximal extremity muscle groups, together with progressive fatigue and malaise, may occur in symptomatic primary hyperparathyroidism. Electromyography changes may be seen in these patients together with atrophy of the skeletal muscle on biopsy specimens.153–155 Although the severe symptoms are rarely encountered, some signs of muscle fatigue and weakness may be present in 40% of patients with mild primary hyperparathyroidism.156 Usually these subtle symptoms are manifested as muscle aches and fatigue on rising from a chair or climbing stairs. Progression of the disease may ultimately result in weakness that limits activity and ambulation over weeks to months. This neuromuscular syndrome is noted to improve after parathyroidectomy in 80% to 90% of the patients affected.157,158
Neurologic System
Neurologic manifestations of primary hyperparathyroidism are represented by a spectrum of symptoms ranging from anxiety and mild emotional disturbance to frank psychosis. Depression, nervousness, and cognitive dysfunction may commonly occur to varying degrees in primary hyperparathyroidism. Cerebral dysfunction characterized by organic brain syndrome is more common in elderly patients with an underlying mild cognitive abnormality exposed to hypercalcemia. Other neurologic changes occasionally seen in patients with hyperparathyroidism include deafness, dysphagia, dysosmia, and dysesthesia.159 Many of the psychiatric symptoms in patients with primary hyperparathyroidism are improved after parathyroidectomy.158,160 Fifty percent of patients with depression or anxiety, or both, improve after surgery. A favorable effect has also been shown in about 50% of patients with organic brain syndrome and dementia. Some older patients experience dramatic improvement; however, it is impossible to predict whether any specific patient will improve after surgery.160
Cardiovascular System
Hypertension may occur in 50% of patients with hyperparathyroidism.161 Convincing evidence of a pathogenic mechanism does not exist, however, and parathyroidectomy results in a reduction in blood pressure in a few of these patients.162 Swedish investigators reported an association with myocardial ischemia and left ventricular dysfunction in patients with hyperparathyroidism and various symptoms, which exhibited reversibility after parathyroidectomy.163
Hypercalcemic Abnormalities
Hypercalcemic syndrome occurring as a result of hyperparathyroidism includes polydipsia and polyuria, anorexia, vomiting, constipation, muscle weakness and fatigue, mental status changes, and skin abnormalities. Patients developing markedly elevated serum calcium levels approaching 15 mg/dL may present with severe mental status changes or coma, a so-called hypercalcemic crisis. If untreated, this condition may progress to acute renal failure and the onset of dysrhythmias, which may precipitate sudden death.164 Other abnormalities include metastatic calcifications at the corneal-scleral junction (so-called band keratopathy), shortened Q-T interval on electrocardiogram, ectopic calcium deposits in various organs, and pruritus. Some patients also may present with a nonspecific debility manifested by anorexia, fatigue, anemia, weight loss, and advancing osteitis, all of which are reversible after parathyroidectomy.
Clinical Course of Untreated Hyperparathyroidism
Several 8- to 10-year prospective studies have shown a benign course of untreated mild hyperparathyroidism in most patients, with no significant progression of symptoms, hypercalcemia, bone marrow density loss, or renal function impairment. No pathologic fractures or kidney stones developed during these observation periods.165,166 There was evidence, however, of disease progression in 27% of patients, including marked hypercalcemia, hypercalciuria, or loss of bone mineral density in 10%.167
Osteitis fibrosis cystica is currently uncommon in patients with primary hyperparathyroidism, and is most often seen in patients with severe hypercalcemia. Bone density measurements have shown an average reduction in cortical bone of 17% among current patients with primary hyperparathyroidism, however, and the bone loss tends to be the most pronounced in postmenopausal women.167–169 Total and trabecular bone mass is often significantly but less markedly reduced.170 No bone loss has been detected in postmenopausal women with borderline hypercalcemia, but losses were significant when the serum calcium level was greater than 2.74 mmol/L.168
Clinically evident renal failure is currently an unusual complication of primary hyperparathyroidism. Reduction of creatinine clearance and urinary concentrating capacity occur in more than one third of patients with mild hypercalcemia, however, indicating that impairment of glomerular and tubular function may occur silently.168 Serum creatinine measurements are crude estimates, and increase only after creatinine clearance is substantially reduced. Serum creatinine levels also decrease with declining muscle mass of aging.168
Rare patients with initially mild hypercalcemia but rapidly advancing disease may have parathyroid carcinoma.171 In addition, stepwise “clinical progression” may occur during observation in patients with primary hyperparathyroidism, possibly representing development of secondary mutations that cause accelerated growth of the tumor. Bleeding in parathyroid tumor may also cause an abrupt increase in serum calcium levels. A history of mild primary hyperparathyroidism has been reported in one third of patients with hypercalcemic crisis. Because it is impossible to predict whether progressive disease will occur in any patient, extended follow-up is crucial if surgery is deferred in primary hyperparathyroidism.172–174
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


