Fig. 1.1
Pierre Robin sequence in a child with Stickler Association
Associations
CHARGE Association
Definition and Clinical Features
From the 1950s to 1970s, case reports began to emerge of children with combinations of coloboma, choanal atresia, and congenital heart disease. Pagon [9] reported an additional 21 cases and coined the moniker “CHARGE association” for the pattern of abnormalities most commonly seen in afflicted patients (Table 1.1) (Fig. 1.2).
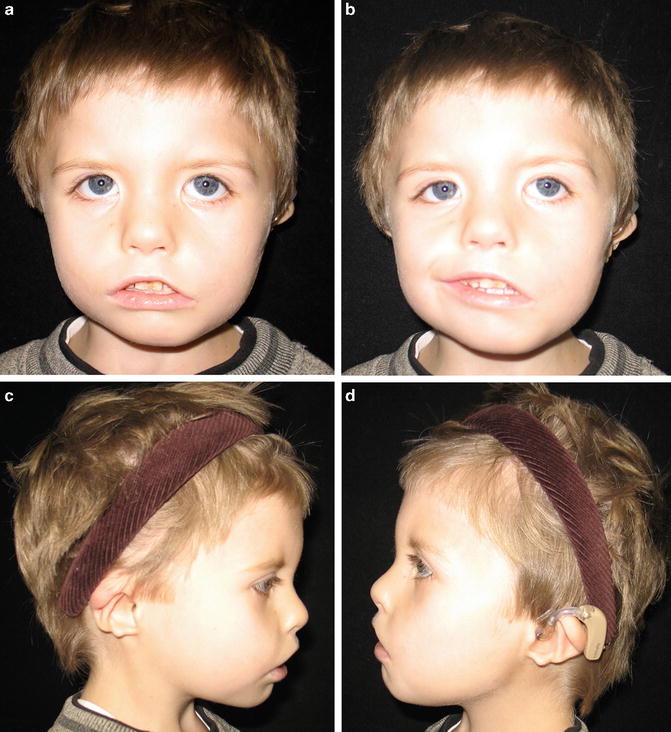
Table 1.1
Clinical features of children diagnosed with CHARGE syndrome
Acronym | Feature | Frequency (%) | Comments |
|---|---|---|---|
C | C = coloboma | 80–90 | Colobomas of iris, retina, or optic disk. May be unilateral or bilateral |
H | H = heart defects | 60–85 | Include patent ductus arteriosum (PDA), ventricular septal defect (VSD), atrial septal defect (ASD), endocardial cushion defect, coarctation of aorta, vascular ring, hypoplastic left heart, and tetralogy of Fallot |
A | A = atresia choanae | 55–85 | May be unilateral or bilateral |
R | R = retardation of growth | 70–85 | Usually normal birth weight with secondary failure to thrive, often falling below the third percentile |
R = retardation of development | 60–100 | Wide range of intellectual impairment, from normal to severe mental retardation | |
G | G = genital defects | 53–100 | Hypogonadism in 80–90 % of males and 15–20 % of females |
E | E = ear anomalies and/or deafness | 85–100 | Characteristic appearance—short, wide, low-set, protruding, lop or cup shaped, with an unusual antihelix and often asymmetric. Mixed progressive hearing loss in 60–90 %. Abnormalities of semicircular canals often noted on computer tomography (CT)/magnetic resonance imaging (MRI) |
Fig. 1.2
Patient with CHARGE Association
Children with a diagnosis of CHARGE typically have at least four of the features, and coloboma or choanal atresia are considered requisite parts of the diagnosis. Other abnormalities seen in CHARGE include structural brain defects [10, 11], cleft lip or palate [12], cranial nerve abnormalities which may be asymmetric [13, 14], esophageal atresia and/or tracheoesophageal fistula, renal abnormalities including horseshoe kidney and ureteral abnormalities [14], and delayed tooth eruption [15].
Heredity and Etiology
CHARGE association results from multiple genetic etiologies (including genetic mutations of CHD7, a gene also known as “chromodomain helicase DNA-binding protein 7,” which is thought to play a role in remodeling chromatin) and based on concordance studies in monozygotic twins and discordance in dizygotic twins is not caused by environmental factors [14, 16, 17]. For children with all major features of CHARGE, CHD7 mutations will be detected in 90 %. For children with suspected CHARGE where some features are absent, a mutation in CHD7 will be identified in fewer patients (60–70 %). Advanced paternal age at the time of conception compared with that of the general population may increase the risk of having a child with CHARGE association. Most cases occur sporadically, and the risk of recurrence in siblings of affected children is about 1 % [18]. There are case reports of children with normal CHD7 sequencing who have mutations in SEMA3E (a member of the semaphorin family of genes that encodes a protein with an immunoglobulin-like domain, a PSI domain, and a Sema domain) [19, 20]. In addition, some children with mutations identified in CHD7 and SEM3AE have also been found to have a deletion at 22q11.2. However, it is likely that additional mutations have not yet been discovered, which may account for the small number of patients with CHARGE who do not have an identified mutation.
Diagnosis
The clinical features of CHARGE are usually identified at birth with some abnormalities evident on prenatal ultrasound examination. In addition to a careful physical exam, a child with suspected CHARGE should undergo hearing and ophthalmologic examination to aid in diagnosis. Head imaging with CT or MRI can identify abnormalities of the semicircular canals as well as structural brain abnormalities. Molecular testing is clinically available and can assist in the diagnosis of CHARGE cases due to CHD7 mutation. Because of the number of cytogenetic abnormalities that can result in a similar phenotype, genomic microarray analysis should be performed for all suspected cases who have normal CHD7 sequencing. Children that are negative for CHD7 mutation can be diagnosed clinically based on the above criteria.
Syndromes
22q11.2 Deletion Syndrome
Definition
The 22q11.2 deletion syndrome (22q11.2 DS) is the most common microdeletion syndrome, with an estimated incidence of 1/4,000 live births [11, 21–26]. Prior to the recognition of its genetic basis, children with different features of the disorder were described with a variety of terms: DiGeorge syndrome, velocardiofacial syndrome, conotruncal anomaly face syndrome, autosomal dominant Opitz/GBBB syndrome, and Cayler cardiofacial syndrome [27–34]. The prevalence of 22q11.2 deletion is even higher, having been identified in 1/68 children with congenital heart disease [35]. This deletion syndrome is the most common cause of syndromic palatal defects and a frequent cause of developmental delay [36].
Clinical Features
Children with 22q11.2 DS have recognizable facial features as well as somatic and functional abnormalities (Fig. 1.3). There are several features that occur frequently in children with 22q11.2 DS, and dozens of other features that have been reported in smaller number of children. The most common abnormalities [37] are listed in Table 1.2.

Fig. 1.3
Patient with 22q11.2 Deletion syndrome
Table 1.2
Clinical features of children diagnosed with 22 q11.2 deletion syndrome
Feature | Frequency (%) |
|---|---|
Developmental delay | >90 |
Congenital heart defect | 76 |
Palatal defects | 76 |
Immunodeficiency | 77 |
Hypocalcemia | 49 |
Renal anomalies | 36 |
Dysphagia | 35 |
Schizophrenia | Approximately 25 |
Polydactyly | 4 |
Congenital diaphragmatic hernia | 1 |
Each of these features occurs at a much greater rate than the general population.
Characteristic facial features in 22q11.2 DS include malar flatness, hooded eyelids, hypertelorism, downslanting or upslanting palpebral fissures, auricular anomalies including overfolded helices, attached earlobes, prominent nasal root with fullness to the nasal tip and hypoplastic alae nasi, nasal dimple or crease, small mouth, and asymmetric crying facies [38–43]. These facial features are variable in any individual child and are less consistently seen in non-Caucasian children [44].
Many other somatic birth defects and functional problems have been reported in children with 22q11.2 deletion, including but not limited to limb defects [45–49], neural tube defects [50], genitourinary anomalies [44], craniosynostosis [39], laryngeal abnormalities [51], autoimmune disorders [52–54], and hearing loss [55].
Heredity and Etiology
The vast majority (85 %) of individuals have the same 2.54 Mb deletion, encompassing about 30 functional genes [37]. Ten percent of cases are familial with a parent usually demonstrating some signs of the disorder. A smaller subset of individuals has smaller deletions within this region [37, 56]. Congenital heart defects in 22q11.2 deletion have been linked with the TBX1 gene (T box transcription factor), as children with smaller deletions that include deletion of TBX1 have a high rate of congenital heart disease [37]. No other clear genotype–phenotype correlations have been made to date [57].
Most deletions are de novo and occur because the structure of the 22q11.2 region is vulnerable to mutations. There are segmental duplications that flank the typical breakpoints, which make it susceptible to rearrangements [58–62]. Like all contiguous gene deletion syndromes, the risk of a child inheriting the deletion from an affected parent is 50 %. However, because of phenotypic variability, the severity of features in a child born to an affected parent cannot be predicted based on the parent’s own phenotype.
Diagnosis
Deletion of the 22q11.2 region can be identified using single-nucleotide polymorphism (SNP) or comparative genomic hybridization (CGH) genome-wide microarray or use of multiplex ligation-dependent probe amplification (MLPA). Although standard fluorescent in situ hybridization (FISH) can be used to detect a deletion of 22q11.2, this technique may miss patients with smaller atypical deletions in 22q11.2 [37, 63–65].
Pregnant women who have a family history of 22q11.2 DS may be offered genetic testing using chorionic villous sampling or amniocentesis. Fetuses with somatic defects noted on ultrasound can be tested utilizing amniocentesis. Sonographic abnormalities identified in pregnancies of fetuses with 22q11.2 DS include congenital heart defects, cleft palate, renal anomalies, polyhydramnios, polydactyly, congenital diaphragmatic hernia, clubfoot, and neural tube defects [44, 57].
Treacher Collins Syndrome
Definition
Treacher Collins syndrome, also known as mandibulofacial dysostosis, is an autosomal dominant disorder with characteristic facial anomalies resulting from malformations of structures arising from the first and second pharyngeal arch, groove, and pouch [66].
Clinical Features (Fig. 1.4)
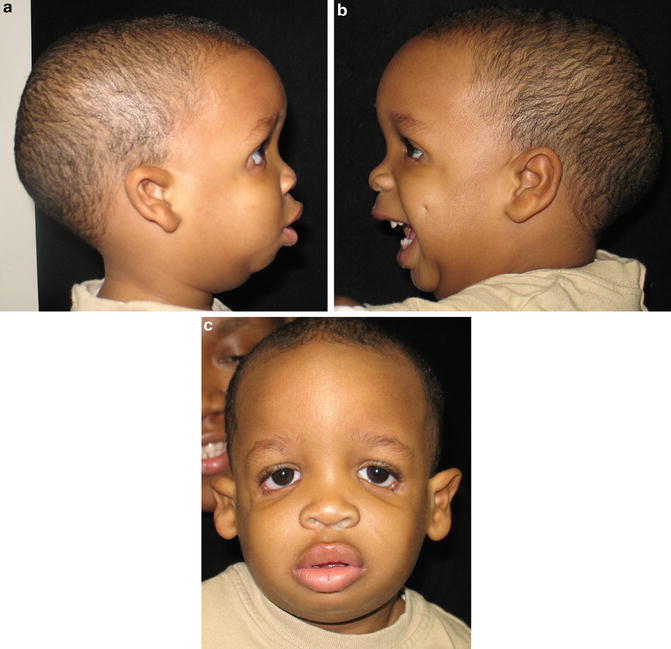
Fig. 1.4
Patient with Treacher Collins syndrome
(a)
Development: Children with Treacher Collins usually display normal intelligence; however, occasional cases of developmental delay have been attributed to hearing loss [67].
(b)
Ears: Malformations of the internal and external ear, include atresia of the external auditory canal, as well as, ossicle abnormalities of the middle ear, and pinnae. Hearing loss is common, secondary to noted defects.
(c)
Eyes: Prominent downslanting of the palpebral fissures; coloboma or notching of the lateral portion of the lower eyelid with absence of eyelashes medial to the notch. Cataracts have also been reported [68].
(d)
Face: Central face appears prominent because of deficiency of zygomatic arch and micrognathia. Micrognathia is the result of mandibular hypoplasia or the condyle, coronoid process, and the body of the mandible with abnormal ramus [66]. The condyle may have abnormalities of the cartilage and is covered with hyaline rather than fibrocartilage.
(e)
Palate: Cleft palate—submucous cleft, often of Robin type (U-shaped cleft with micrognathia and glossoptosis). Cleft lip is rare.
(f)
Skull: Skull anomalies including brachycephaly with bitemporal narrowing, absent or underdeveloped malar bones with nonfusion of the zygomatic arches, hypoplastic zygomatic process, lateral pterygoid plates and muscles, non-pneumatized mastoids, small or absent paranasal sinuses, and absence of infraorbital foramen.
(g)
Other characteristic features: Brachycephaly, tongue-shaped extension of hair protruding in front of ear, preauricular ear tags, or fistulae externalizing between tragus and corner of mouth.
Heredity and Etiology
Treacher Collins is autosomal dominant with variable penetrance. Approximately 40 % of cases are familial. Mutations of TCOF-1, also known as Treacle, are identified in 70–93 %. Somatic mosaicism has been reported [69–72]. Treacle mutations lead to haploinsufficiency of a nucleolar phosphoprotein involved in ribosomal RNA production. Animal models of Treacher Collins have been produced by inducing haploinsufficiency of this gene. In mice, haploinsufficiency of Treacle is associated with apoptosis of neural crest cells and prefusion of the neural folds, although the mechanism of these events remains unclear. Occasional cases of Treacher Collins with normal TCOF–1 have been associated with mutations of polymerase (RNA) 1 polypeptide C (POLR1C) [73]. Exposure to isotretinoin in utero can also reproduce craniofacial abnormalities similar to Treacher Collins.
Associated Malformations
Treacher Collins has clinical overlap with Nager syndrome, Miller syndrome, and Robin sequence.
Craniosynostosis Syndromes
Definition
Craniosynostosis, the premature fusion of cranial sutures, occurs in approximately 1/3,000 births. It is most often an isolated finding, affecting the sagittal or coronal sutures. Craniosynostosis can also occur as part of a syndrome, with additional findings such as limb defects and developmental delay. The most common craniosynostosis syndromes are Apert syndrome, Crouzon syndrome, Pfeiffer syndrome, Muenke syndrome, and Saethre–Chotzen syndrome, although more than 100 syndromes associated with craniosynostosis have been described. Figures 1.5, 1.6, and 1.7 demonstrate the typical facial features of children with Apert, Pfeiffer, and Crouzon syndrome respectively.
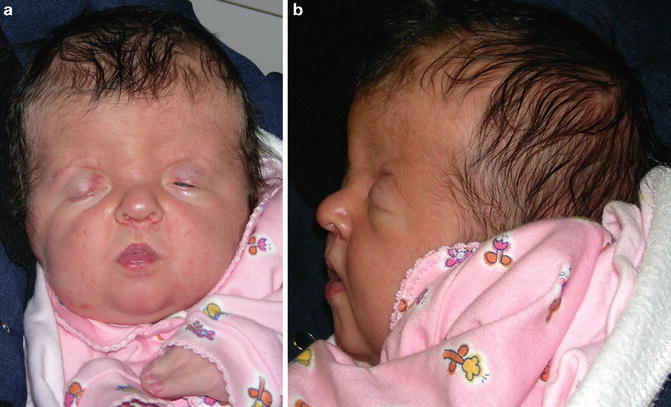
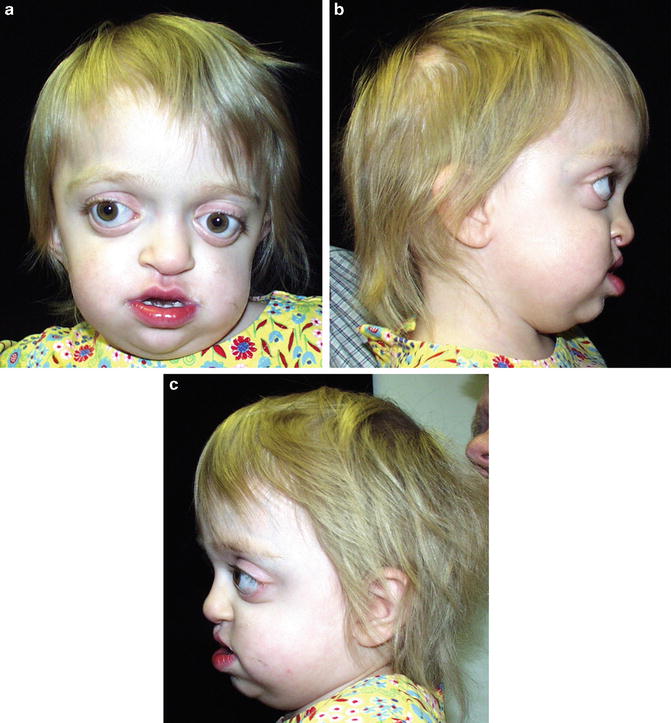
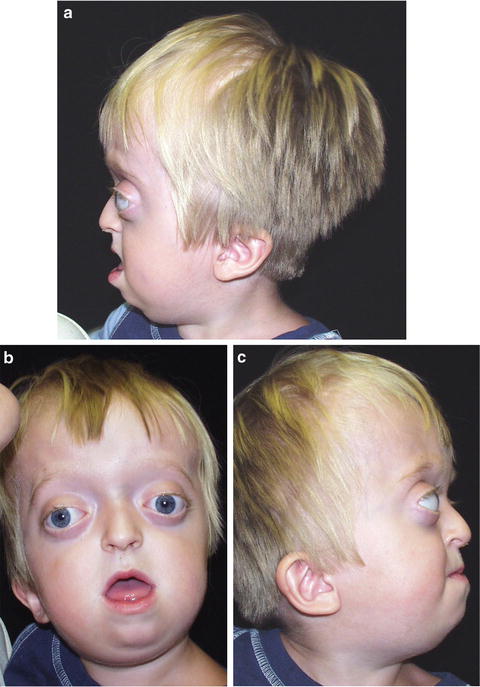
Fig. 1.5
Infant with Apert syndrome
Fig. 1.6
Patient with Pfeiffer syndrome
Fig. 1.7
Patient with Crouzon syndrome
Clinical Features
Apert syndrome: Apert syndrome is easily distinguished from other craniosynostosis syndromes by the presence of bilateral severe syndactyly of the hands and feet. The thumb is typically spared, with most severe involvement of the second through fourth fingers. This gives the hand the appearance of a mitten. The feet typically involve all toes, with relative sparing of the hallux. The skull has a short anterior–posterior diameter which causes the child to have a high forehead due to bicoronal synostosis. There are often irregularities of the synostosis. Children with Apert syndrome have typical unusual facial features including supraorbital grooves, ocular hypertelorism and downslanting palpebral fissures, depressed nasal bridge with short, broad nose, and long upper lip with thin vermillion border [79]. Most affected individuals have cognitive impairment. Other clinical features in Apert include cardiac defects (10 %), genitorurinary abnormalities (10 %), cleft palate (43 %), choanal stenosis, agenesis of the corpus callosum, and ventriculomegaly [79–81].
Crouzon syndrome: Children with Crouzon syndrome also typically have bicoronal synostosis but lack involvement of the hands or the feet. Facial features include hypertelorism, proptosis, midfacial hypoplasia, and a beaked nose. Occasionally, radiographic abnormalities of the carpal bones may be seen. Children with Crouzon do not typically have involvement of other organ systems, and cognitive development is normal [79].
Pfeiffer syndrome: Children with Pfeiffer syndrome can be distinguished from those with other craniosynostosis syndromes because they also have broad and medially deviated great toes and broad thumbs. The craniosynostosis in these children results in bicoronal synostosis, but they may also have deformities that result in a cloverleaf-shaped skull. The severity of the phenotype can vary in children with Pfeiffer syndrome, with some being severely affected. Children with Pfeiffer syndrome should also be evaluated for choanal stenosis and tracheal sleeve.
Muenke syndrome: Muenke syndrome is characterized by bicoronal or unicoronal synostosis. Macrocephaly, hearing loss, and brachydactyly have also been reported. Abnormalities of the hands, such as thimble-shaped phalanges, may be seen radiographically. There are mild learning disabilities and variable hearing loss associated with this syndrome [79].
Saethre–Chotzen syndrome: Although Saethre–Chotzen is a craniosynostosis syndrome, the presence of craniosynostosis is not an obligate finding for diagnosis. There are a variety of minor clinical features that can be seen in family members, which may lead to genetic testing and diagnosis. There is variable presence and involvement of cranial sutures. The coronal sutures are typically involved in Saethre–Chotzen, but other sutures may be affected resulting in skull and facial asymmetry. Ocular abnormalities in Saethre–Chotzen include ptosis and tear duct stenosis. The ears have characteristic findings including prominent crus and helical roots. The hands are often short and have a single palmar crease and mild syndactyly. The feet have broad or bifid halluces and syndactyly of the toes, which deviate laterally, though not as severely as seen in those children who have Apert syndrome. The cognitive development of these children is normal in most individuals.
Heredity and Etiology
Most craniosynostosis syndromes, including those discussed here, are caused by mutations in FGFR1, FGFR2, FGFR3 (fibroblast growth factor genes), and TWIST (a transcription gene affecting DNA binding and helix–loop–helix domains).
Apert syndrome: Apert syndrome is caused by one of two point mutations (S252W or P253R) in FGFR2 leading to an amino acid change [82]. This leads to increased signaling activity due to reduced dissociation of the receptor/ligand complex. Apert syndrome is transmitted autosomal dominantly with complete penetrance, but most cases represent new mutations, and this is reflected in that the affected individuals are more severely affected. The incidence of Apert syndrome increases with advanced paternal age.
Crouzon syndrome: Crouzon syndrome has been associated with a variety of mutations in FGFR2. In addition, there is some overlap of mutations of genes that cause Crouzon syndrome and other craniosynostosis syndromes. These mutations are thought to be “gain-of-function mutations” because they confer new or enhanced activity on proteins such as the fibroblast growth factor receptor 2 protein as seen in Crouzon syndrome; however, about half of the cases of Crouzon are not a result of an identified mutation. Inheritance is autosomal dominant, and many cases are familial as there is no effect on reproductive fitness.
Pfeiffer syndrome: Like Crouzon, numerous mutations in FGFR2 can lead to Pfeiffer syndrome. All mutations in familial cases of Pfeiffer are thought to be autosomal dominant. Mutations are generally de novo in severe cases where reproductive fitness is reduced. Occasional mild cases with mutation in FGFR1 have been noted.
Muenke syndrome: Muenke syndrome is caused by a Pro250Arg mutation in FGFR3. This too is thought to be a “gain-of-function mutation” and is transmitted in an autosomal dominant pattern, and the syndrome may be unrecognized in mildly affected family members.
Saethre–Chotzen syndrome: Saethre–Chotzen is caused by mutations in TWIST, a developmentally regulated transcription factor [83–85]. A translocation at the TWIST locus has also been reported to result in Saethre–Chotzen [86]. Deletion of TWIST can also lead to Saethre–Chotzen. Inheritance of Saethre–Chotzen is autosomal dominant.
Diagnosis
The diagnostic evaluation of children with craniosynostosis should begin with a careful physical exam and attention to associated features as there is molecular overlap in some of the craniosynostosis-related syndromes. Clinical testing is available for FGFR1, FGFR2, FGFR3, and TWIST. Special considerations and individual testing recommendations are given below:
Apert syndrome: FGFR2 sequencing only.
Crouzon syndrome: FGFR2 sequencing should be done, but mutations are only identified in 60 % of cases, so a negative result does not rule out a diagnosis.
Pfeiffer syndrome: FGFR2 sequencing is recommended. If negative, consider sequencing of FGFR1. Sequencing of FGFR1 can also be considered as part of the original work-up in mild cases.
Muenke syndrome: FGFR3 sequencing for Pro250Arg mutation.
Saethre–Chotzen syndrome: TWIST sequencing and microdeletion analysis. If these results are normal and the diagnosis is suspected, chromosome analysis for balanced translocations should be performed.
Oculo-Auriculo-Vertebral Spectrum (Hemifacial Microsomia, Goldenhar Syndrome)
Definition
Goldenhar syndrome, part of the oculo-auriculo-vertebral spectrum (OAVS), is characterized by variable degrees of underdevelopment of the ear, mouth, and mandible of one or both sides of the face as well as vertebral anomalies, epibulbar dermoids, cleft palate, and/or hearing loss. Hemifacial macrosomia, in which no epibulbar dermoids are noted, is also part of the OAVS. This group of disorders has also been termed “first and second branchial arch syndrome” based on the embryologic etiology [87–90]. Other disorders in the OAVS include craniofacial microsomia, Goldenhar–Gorlin syndrome, first arch syndrome, facio-auriculo-vertebral syndrome, and lateral facial dysplasia. The distinction of specific disorders along the OAVS has been an area of debate, but cases classified as Goldenhar are thought to represent 10–35 % of cases. The incidence of OAVS is estimated to be 1/5,600 [91]. The male:female ratio is approximately 3:2.
Clinical Features
There is considerable variability in the type and severity of abnormalities in OAVS. Facial features of hemifacial microsomia can be seen in Fig. 1.8, and features typical of Goldenhar can be seen in Fig. 1.9. The phenotypic appearance of the facial findings has been used for further classification within the spectrum, but many other organ systems can also be involved.
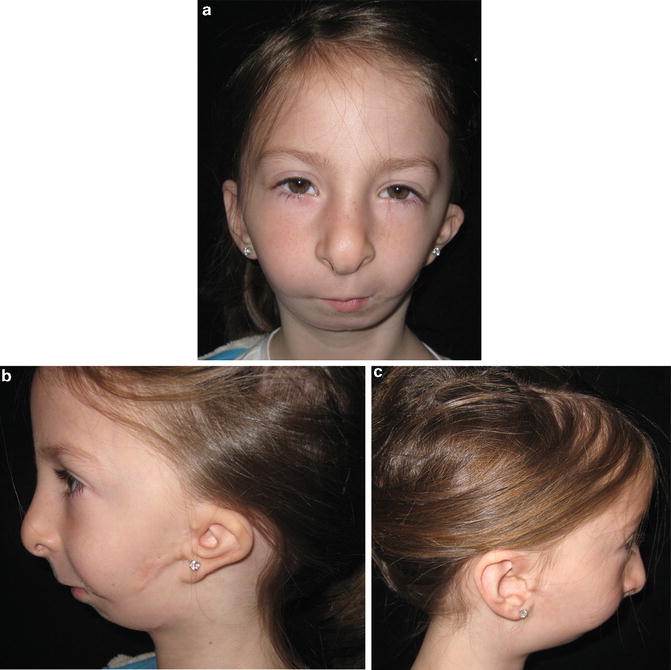
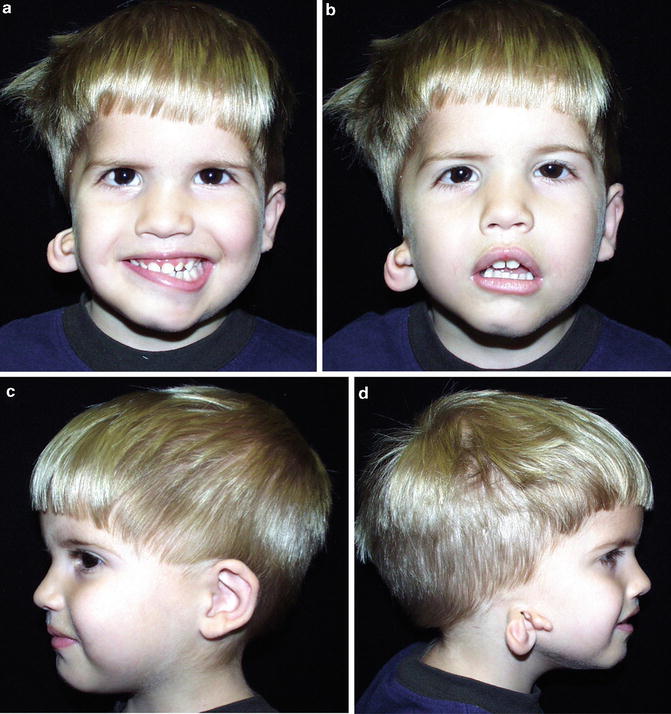
Fig. 1.8
Patient with Hemifacial microsomia
Fig. 1.9
Patient with Goldenhar syndrome
(a)
Facies: Facial asymmetry is seen in the majority of cases, even in children who may have bilateral involvement. Mild cases are often not noticed in infancy but are usually obvious by the age of four [87]. The appearance of asymmetry is produced by a combination of abnormalities of the bony structures, musculature, and soft tissue. Kaban et al. [92] published a revised classification of this disorder based on facial bone abnormalities:
Type I, miniature mandible with normal morphology
Type IIA, mandibular ramus abnormal in size and shape
Type IIB, mandibular ramus abnormal in size, and shape and location requiring costochondral graft construction
Type III, absent ramus, condyle, and temporomandibular joint
In all cases, the temporal bone, orbit, zygoma, nasal bones, and maxilla may be distorted. Those with types IIB and III have the most severe distortion, and affected children often have absent zygomatic arch and hypoplastic or inferiorly displaced orbit.
(b)




Eyes: A variety of ocular abnormalities are evident. Epibulbar dermoids are the most common ocular abnormality, occurring in up to 35 % of individuals with OAVS [93, 94]. Epibulbar dermoids are white or yellow solid masses that appear on the globe or the orbit, most commonly on the inferotemporal quadrant at the limbus. Typically, children with epibulbar dermoids are classified as having Goldenhar syndrome. Bilateral lesions occur in 25 % of patients and often appear at the same location in each eye. Vision may be impaired as a result of obstruction of the papillary axis, astigmatism, or lipid infiltration of the cornea. Other structural ocular abnormalities may include unilateral colobomas of the upper lid (20 %), bilateral colobomas (3 %), blepharoptosis (narrowing of the palpebral fissure) (10 %), elevation of the orbit (7 %) [95], and, in severe cases, anophthalmia and microphthalmia [93, 94, 96–100]. Patients with epibulbar dermoids have a higher rate of other ocular abnormalities such as blepharoptosis, microphthalmia, and anophthalmia [93, 101, 102]. Ocular motility defects are seen in 25 % of cases, including Duane syndrome, esotropia, and exotropia.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
