Fig. 2.1
Schematic demonstrating the hillocks of His that develops into the mature auricle (reprinted with permission from Moore KL, Persaud TVN. The Developing Human: Clinically Oriented Embryology. 5th ed. Philadelphia: W.B. Saunders; 1993:438)
The EAC develops from the dorsal portion of the first pharyngeal groove, which progressively deepens during the second month (Fig. 2.2). The ectoderm of the groove eventually abuts the endoderm of the tubotympanic recess, which is derived from the first pharyngeal pouch. This contact is brief and at the sixth month is broken by a mesodermal ingrowth. At 8 weeks, the inferior portion of the first pharyngeal groove deepens again. This forms the primary EAC, which corresponds to the cartilaginous canal in the adult. At 9 weeks, a cord of epithelial cells at the bottom of the primary EAC grows medially into the surrounding mesenchyme to terminate in a solid epithelial plate, which is known as the meatal plug. It is not until after the fifth month that the plug splits open, initially at its medial terminus, forming the bony EAC by the seventh month. The cells remaining at the periphery form the epithelial lining of the bony EAC, whereas those remaining medially form the superficial layer of the tympanic membrane (TM). These developmental changes in the EAC occur at a time when the outer, middle, and inner ear are already well developed.
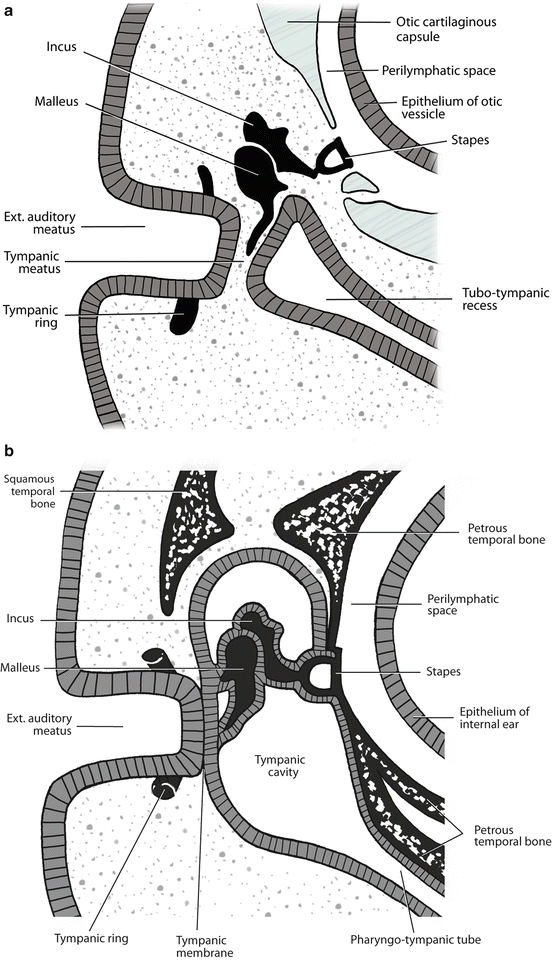
Fig. 2.2
Embryology of ear canal
The TM is trilaminar. The primordium of the TM is the pharyngeal membrane, which separates the first pharyngeal groove from the first pharyngeal pouch. At 9 weeks, the mesenchyme adjacent to the meatal plug gives rise to the lamina propria of the TM. This mesenchyme is surrounded by the four ossification centers of the tympanic ring. By the tenth week, the tympanic ring elements fuse except for a small region superiorly where a defect remains; this becomes the notch of Rivinus. These elements then expand, accompanied by the growth of the solid epithelial cord of cells of the meatal plug. Thus, the TM develops from three sources that correspond to its trilaminar structure. Its lateral surface arises from ectoderm of the first pharyngeal groove; its medial surface from endoderm of the tubotympanic recess, a derivative of the first pharyngeal pouch; and its middle layer, a fibrous stratum of connective tissue, from mesenchyme of the first and second pharyngeal arches.
Preauricular Sinus
Definition
A preauricular sinus is a congenital malformation that manifests as an external small opening in the preauricular region. It has been variably termed preauricular pit, preauricular fistula, preauricular tract, and preauricular cyst.
Incidence
Etiology
The most accepted theory attributes the development of a preauricular sinus to incomplete or defective fusion of the six hillocks of His [10].
Associated Malformations
The preauricular sinus has been described as part of a number of syndromes, most commonly branchio-oto-renal (BOR) syndrome [14, 15]. Others include brancho-oto-costal syndrome [16], cat eye syndrome [17], Waardenburg’s syndrome [18], and trisomy 22 [19, 20].
Some studies have found an association between hearing loss (conductive and sensorineural) in newborn infants with isolated preauricular tags and pits. This incidence has been reported as high as 15–30 % [21]. Interestingly, the hearing loss may evolve after a few years. However, many studies have found no statistically significant associated hearing loss with isolated preauricular sinuses and tags compared to control groups [10, 22]. The incidence of external or middle ear anomalies does not appear to be higher in patients with an isolated preauricular sinus. Conversely, individuals with congenital branchial cleft anomalies may have a higher incidence of concomitant preauricular sinus, reported as high as 60 % in the literature [23]. It is well known that patients with syndromic preauricular sinuses may have associated hearing loss as part of the clinical spectrum [24, 25].
Studies have shown an association between preauricular sinus and renal structural abnormalities. The percentage of children with isolated preauricular sinus and associated major renal structural abnormalities is reported to be 2.2–4.3 % [14, 26]. A prospective study of over 32,000 live births suggested a slightly increased risk (odds ratio 1.3) of renal anomalies in children with ear anomalies [21].
Clinical Features
Over 50 % of cases overall are unilateral [27], although some studies have found the incidence of bilateral lesions to be 25–50 % of cases [9]. They occur more commonly on the right side [8, 10]. They are frequently noted during routine physical examination as a small pit adjacent to the external ear, usually located at the anterior margin of the ascending limb of the helix, although the opening has also been reported along the posterosuperior margin of the helix, the tragus, or the lobule [8, 28]. The extent of the depth of the pit may vary, and can be shallow, limited to the visible portion of the pit, or may form a sinus tract that can extend to a variable depth or branch or follow a tortuous course. A preauricular sinus may also result in the formation of a subcutaneous cyst. These cysts are intimately related to the tragal cartilage and the anterior crus of the helix, whereby they are always adherent to the perichondrium of the auricular cartilage [29]. Unlike the tract of first branchial arch anomalies, a preauricular sinus tract is lateral and superior to the facial nerve and parotid gland.
Patients may present with discharge from the sinus either as a result of desquamating epithelial debris or infection. The latter may manifest with erythema, swelling, pain, and discharge. The most common culture pathogens implicated in infection are Staphylococcal species and, less commonly, Proteus, Streptococcus, and Peptococcus species [9].
Classification
A preauricular sinus can be inherited or can occur sporadically. Most cases are sporadic. Bilateral cases are more likely to be inherited [9]. When inherited, the pattern is of incomplete autosomal dominance with reduced (around 85 %) penetrance [27, 30]. Research studies have mapped a possible locus for congenital preauricular fistula to chromosome 8q11.1–q13.3 [31].
Diagnosis
Most patients are asymptomatic, and a sinus pit in a typical site is highly suggestive of the diagnosis (Fig. 2.3). A thorough history and head and neck examination are mandatory in all cases, seeking evidence of associated anomalies that may lead to the diagnosis of a syndromic etiology. Assessment of bilateral involvement, facial nerve function, and external ear development is important.
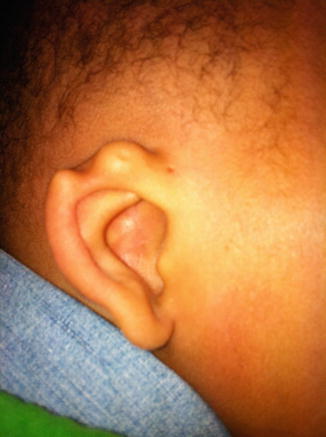
Fig. 2.3
Child with two ear pits (the more common preauricular pit and a less common helical pit adjacent to the notch in the helix)
Management
Assessment of a child with a preauricular sinus, pit, or tag entails a thorough head and neck examination as well as systems review to elicit any obvious syndromal association. Assessment of the auricle as well as otomicroscopic examination of the external auditory canal and tympanic membrane are carried out to assess for any concomitant outer ear malformations.
The necessity of performing a renal ultrasound in patients with an isolated preauricular sinus has been controversial. Some authors have recommended that renal ultrasounds be performed on all patients with isolated preauricular sinuses [14], although this view is not shared by others [28]. Compelling arguments for attaining a renal ultrasound include the low cost and risk associated with the test as well as the significant clinical implications of a detected renal anomaly. Wang et al. [21], based on a retrospective review of 42 children with external ear anomalies who had undergone renal ultrasound in two genetics medical centers between 1981 and 2000, suggested that renal ultrasound should only be performed on patients with a preauricular sinus and one or more of the following:
1.
Another malformation or dysmorphic feature
2.
A family history of deafness
3.
An auricular and/or renal malformation
4.
A maternal history of gestational diabetes
Routine audiometric testing in patients with an isolated preauricular sinus has also been controversial. Most of the conducted studies constituted small patient cohorts. Moreover, confounding factors, including otitis media with effusion in the young age group, make data interpretation difficult. There has been no definitive evidence that hearing assessment should be carried out in the routine evaluation of the newborn with an isolated preauricular sinus, at least no more so in these children than in those without preauricular pits.
Management of an acutely infected preauricular sinus entails administration of appropriate antibiotics active against the causative pathogen. In the case when an abscess is present, incision and drainage are commonly undertaken. This can however result in scarring that may make subsequent definitive surgical excision of the sinus more challenging. Coatesworth et al. [32] have described a technique for drainage of a preauricular abscess using a lacrimal probe, in an attempt to minimize trauma to the underlying sinus, potentially reducing the impact of fibrosis on subsequent surgical excision if needed.
Management of recurrent or persistent preauricular sinus infections usually requires surgical excision of the sinus and its tract, preferably at a time of quiescence. Various surgical techniques have been described, with the primary surgical goal of obtaining complete excision to reduce the risk of recurrence. In reviewing the literature, recurrence rates have been reported between 0 and 42 % [33, 34]. The classic approach to surgical excision involves an elliptical skin incision with removal of the skin and sinus opening and medial dissection of the sinus tract. Wide local excision in the form of a radical supra-auricular approach has also been described [34]. This approach involves a postauricular extension of the elliptical incision around the sinus opening, with subsequent medical dissection to the temporalis fascia and continued over the cartilage of the anterior helix. Tissue superficial to the temporalis fascia is removed together with the preauricular sinus as well as a portion of the cartilage or perichondrium of the helix at the base of the sinus. Studies have shown that the supra-auricular approach has a lower recurrence rate compared with simple sinus and tract excision (3.7–5 % vs. 32–42 %, respectively) [35, 36].
Currie et al. [33] carried out a retrospective review over a period of 8 years to assess risk factors that influenced recurrence rates following surgical excision of preauricular sinuses. Factors found to reduce the risk of recurrence were the use of the supra-auricular approach with clearance down to the temporalis fascia, avoidance of sinus rupture, and closure of wound dead space. To facilitate complete surgical excision, many techniques have been employed to aid in the visualization of the sinus tract and its branches. These include preoperative sonographic imaging, preoperative sinograms, intraoperative methylene blue injection, and the use of a lacrimal probe [37, 38]. These have proven to be of variable benefit, and their use is mostly dictated by surgeon preference.
Aural Atresia
Definition
Congenital aural atresia (CAA) refers to a spectrum that involves varying degrees of failure of development of the EAC.
Incidence
The prevalence rates reported for CAA have been variable, reasons for which include variable registration systems and lack of a standardized definition and diagnosis [39]. Furthermore, CAA is often associated with microtia; accurately capturing CAA data from this overlapping dataset has been difficult. According to the Swedish Board of Welfare statistics, the frequency of isolated EAC malformations in 1980 was reported as 0.92 per 10,000 live births.
Etiology
Failure of canalization of the EAC canal during the seventh gestational month results in membranous and/or bony CAA. The etiology of CAA, as with microtia, is multifactorial. A number of factors have been implicated in the pathogenesis, including teratogens, vascular insults, and genetic aberrations [40, 41].
Associated Malformations
Microtia is commonly associated with CAA, and the degree of auricular malformation usually correlates with the degree of CAA as well as the middle ear deformity. However, even though segments of the auricle, EAC, and ossicular chain originate from the same pharyngeal arches, they are from different segments of neural crest cells and evolve at different times in embryonic development. As such, a severe microtia with accompanying CAA is not always accompanied by a disruption in the development of the ossicular chain or middle ear cavity. Likewise, cases of a normal auricle with CAA or, albeit rarely, of microtia with normal EAC and middle ear cavity have been reported [42]. Given the separate embryologic origin of the inner ear, the incidence of inner ear abnormalities is relatively low in patients who have CAA (about 8 %) [43]. CAA has been reported in patients with chromosomal anomalies, especially terminal deletions at chromosome 18q23. A study by Veltman et al. found that atresia occurs in approximately 66 % of all patients who have a terminal deletion of 18q [44].
Clinical Features
CAA is more commonly unilateral (70–90 %) [45]. When bilateral, the deformity on either side can vary in complexity. For unknown reasons, males are affected more commonly than females, at a 2.5:1 ratio. In addition, the right ear is more commonly involved (~60 %) [46]. CAA is more often bony than membranous, and it can be complete or incomplete. The latter may result in a stenotic cartilaginous canal laterally, with a medial osseous canal of normal caliber and a normal TM.
Classification
Several classifications have been proposed based on various parameters including clinical, radiological, surgical, and histopathological findings. A commonly used classification of CAA is that described by Weerda [47], which categorizes these malformations into three types (Table 2.1).
Table 2.1
Weerda classification of congenital aural atresia
Type A | Marked narrowing of the EAC with an intact skin layer |
Type B | Partial development of the EAC with an atresia plate at the medial part |
Type C | Complete bony EAC atresia |
Altmann was the first to describe a histopathological classification correlating the severity of CAA [48] (Table 2.2). He divided three categories: mildly, moderately, and severely malformed types. Many authors have since modified this classification system, further subclassifying type II based on the surgical findings and functional outcome [49] (Table 2.3).
Table 2.2
Altmann classification of congenital aural atresia
Group 1 (mild) | Some part of the EAC, although hypoplastic, is present. The tympanic membrane is hypoplastic, and the eardrum is small. The tympanic cavity is either normal in size or hypoplastic |
Group 2 (moderate) | The EAC is completely absent, the tympanic cavity is small and its contents deformed, and the “atresia plate” is partially or completely osseous |
Group 3 (severe) | The EAC is absent, and the tympanic cavity is markedly hypoplastic or missing |
Table 2.3
Modified Altmann classification of CAA
Type I: Mild | Tympanic membrane is smaller in area than normal; various kinds of ossicular malformations may be present |
Type II: Moderate | Moderate: atretic plate; tympanic cavity is within normal limits |
IIa | Tympanic bone hypoplastic; course of the facial nerve usually normal |
IIb | Tympanic bone absent; abnormal course of the facial nerve |
Type III: Severe | No canal, and a severely hypoplastic tympanic cavity |
Schuknecht described a classification system (types A–D) based on a combination of high-resolution computed tomography (CT) scan and surgical findings (Table 2.4) (Fig. 2.4a–d).
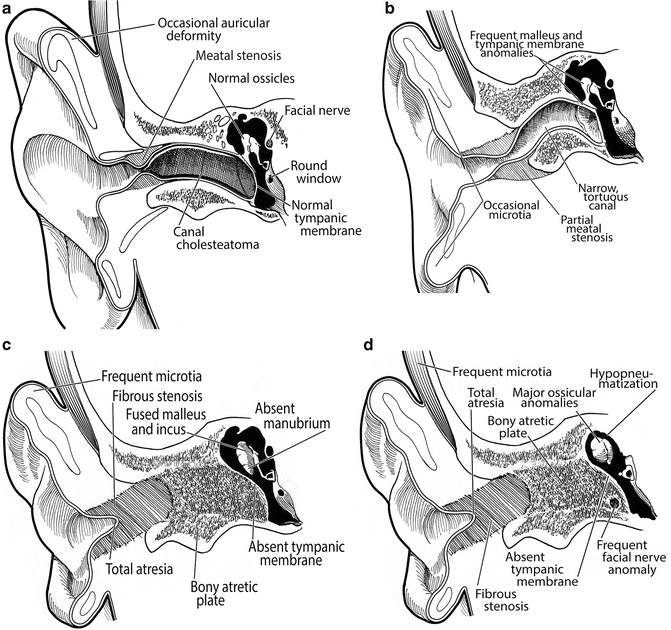
Table 2.4
Schuknecht classification of CAA
Type A: Meatal atresia | Limited to a stenotic fibrocartilaginous portion of the EAC |
Type B: Partial atresia | Narrowing and sometimes tortuousities of both the fibrocartilaginous and bony parts of the EAC. The TM size is often reduced and replaced at least in part by a bony septum, and there is usually middle ear deformities including ossicular malformations |
Type C: Total atresia | Totally atretic canal, but well-developed pneumatization of the tympanic cavity. Characteristically, the TM is missing, the head of the malleus is slightly deformed with fixation to the atresia plate, and the head of the malleus and body of the incus are fused. The facial nerve occasionally takes a more anterior course, in which case it may partially overlap the oval window |
Type D: Hypopneumatic total atresia | Displays the same dysmorphic features found in type C as well as reduced pneumatization of the temporal bone. There is usually an abnormal course of the facial nerve as well as abnormalities of the bony labyrinth |
Fig. 2.4
(a–d) Schematics of types of ear canal stenosis/atresia (based on Schuknecht classification system)
Diagnosis
CAA is typically evident on initial neonatal examination. The presence of microtia may draw attention to a possible associated CAA. In many countries and in all states within the United States, universal newborn hearing screening is mandatory. This has enhanced the early identification of children with more subtle presentations of CAA (e.g., unilateral CAA) who might not have been diagnosed until later in life.
In the absence of other major congenital malformations, it is most important to evaluate the status of the child’s hearing. Even if the contralateral ear is normal on newborn screening, a diagnostic auditory brainstem response (ABR) test is typically recommended in order to assure that the child has at least one normal hearing ear for access to sound and speech. Aside from audiometric assessment, any patient with CAA in whom surgical repair is contemplated should undergo high-resolution CT scanning of the temporal bones (Fig. 2.5).
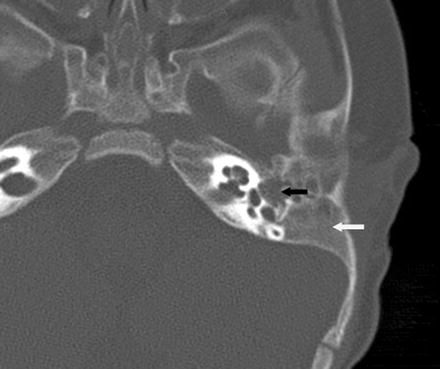
Fig. 2.5
Axial computed tomography of left temporal bone demonstrating auricular atresia with absence of an ear canal (white arrow) and malformed middle ear space (black arrow)
Management
In cases of unilateral CAA, hearing is typically normal in the contralateral ear [50]. Some syndromes, such as Goldenhar syndrome or hemifacial microsomia, present an important exception to this, because the uninvolved ear may demonstrate a conductive, sensorineural, or mixed hearing loss. CAA typically results in conductive hearing loss (CHL) in 80–90 % of the cases with the remaining patients demonstrating a sensorineural hearing loss (SNHL) component [5]. The CHL is typically in the moderate hearing loss range of 40–60 dB.
Current literature suggests that there are many benefits to binaural stimulation, and children with a unilateral hearing loss are at a greater risk of delayed language development, attention-deficit disorders, and poor school performance. As such, patients with unilateral CAA are currently managed with various rehabilitative options, including a hearing aid, assistive listening devices, as well as surgical correction of the atresia. In patients with complete CAA who opt to use a hearing amplification device, a bone-conduction hearing aid must be used, while in cases of incomplete CAA, an air-conduction hearing aid is also an option, but can be problematic due to difficulties in effectively fitting a stenotic ear canal. In the classroom setting, an FM system can also be an extremely effective option for children with CAA and unilateral hearing loss.
In bilateral CAA, the degree of hearing loss is usually significant enough to cause delay in speech and language acquisition and overall psychological development. It is recommended that the medical and audiologic evaluation and management of infants with CAA should follow the American Academy of Pediatrics 1–3–6 newborn hearing loss guidelines: screen by 1 month, diagnose by 3 months, and begin intervention by 6 months of age.
If surgical correction is contemplated, preoperative CT scanning provides valuable information to help in the decision-making process. In 1992, Jahrsdoerfer et al. [51] proposed a CT grading system for CAA that was shown to correlate with postoperative hearing outcomes (Table 2.5). With respect to surgical management of unilateral CAA, the risks of atresia repair should be carefully discussed with the family and weighed against the alternatives of a bone- or air-conduction hearing aid or implantation of a bone-anchored hearing aid. CAA repair carries the risks of canal restenosis, facial nerve injury, sensorineural hearing loss, and postoperative chronic otorrhea. The degree of audiometric improvement is also somewhat unpredictable following atresia surgery. Delaying surgery until after adolescence, when patients can share in their own decision-making process, is advocated by some otologic surgeons. The goal of surgery in patients with bilateral CAA is to restore sufficient hearing such that amplification is no longer needed. This typically translates to a hearing threshold of 25 dB or better. The choice of which ear to operate on is based on imaging evaluation rather than audiometric assessment. Using the Jahrsdoerfer CT grading system, in cases of unilateral CAA, a good surgical candidate scores 8 or more points. In bilateral CAA, the better ear (higher score) is chosen for repair first. With proper patient selection in cases of aural atresia, it is possible to achieve a hearing level of 25 dB or better in 50–70 % of patients. If microtia is present, its repair is first carried out as the child approaches school age (typically 5 years of age). CAA repair follows thereafter at about 6–7 years of age.
Table 2.5
Jahrsdoerfer grading system of candidacy for CAA repair
Parameter | Points |
|---|---|
Stapes present | 2 |
Oval window open | 1 |
Middle ear space | 1 |
Facial nerve normal | 1 |
Malleus–incus complex present | 1 |
Mastoid well pneumatized | 1 |
Incus–stapes connection | 1 |
Round window normal | 1 |
Appearance of external ear | 1 |
Rating | Type of candidate |
|---|---|
10 | Excellent |
9 | Very good |
8 | Good |
7 | Fair |
6 | Marginal |
≤5 | Poor |
The risk of cholesteatoma development with CAA is about 4–7 % [40]. A CT scan of the temporal bones should routinely be obtained around 4 years of age. It is not recommended that a CT scan be obtained at an earlier age, as it would have to be repeated closer to the time of surgery given the ongoing growth and maturity of the temporal bone. Furthermore, the risk of cholesteatoma development under 3 years of age is minimal. This approach further minimizes unnecessary radiation exposure in the pediatric population. If cholesteatoma does exist, surgery is planned, in the form of either canal and middle ear surgery if the CT scan is favorable or canaloplasty alone.
Microtia
Definition
Microtia describes a spectrum of malformations that span a wide range of clinical presentations that can affect the size, orientation, position, and morphology of the auricle. Complete absence of the auricle can also occur (anotia).
Incidence
Etiology
The etiology of microtia is often multifactorial. Only 15 % of patients have a positive family history. In a minority of patients, a genetic or an environmental cause can be found; in these cases, microtia is usually part of a specific pattern of multiple congenital anomalies.
Associated Malformations
Microtia is commonly associated with CAA, and the degree of auricular deformity correlates with the degree of middle ear deformity and conductive hearing loss. Microtia occurs in association with several single-gene disorders, such as Treacher Collins syndrome, as well as chromosomal syndromes, such as trisomy 18. It is frequently associated with the oculoauriculovertebral dysplasia spectrum of congenital anomalies, including Goldenhar–Gorlin syndrome [53]. Furthermore, microtia is a component of isotretinoin and thalidomide teratogenicity and can be part of fetal alcohol syndrome and maternal diabetes embryopathy [33]. Women with four or more pregnancies are at increased risk for having a child with microtia, especially its most severe form (anotia) [54].
Clinical Features
Classification
Many classification systems have been proposed and modified over the years. In essence, all of the classification systems classify a normal or near-normal auricle as Grade I, with increasing grades signifying increasing deformity. Commonly used classification systems include the De la Cruz and Weerda classification systems. The De la Cruz classification divides malformations into minor and major categories (Table 2.6). In 1988, Weerda compiled all the classification systems into one scheme that is useful for clinical grading as well as basic management principles (Table 2.7) (Fig. 2.6a–c).
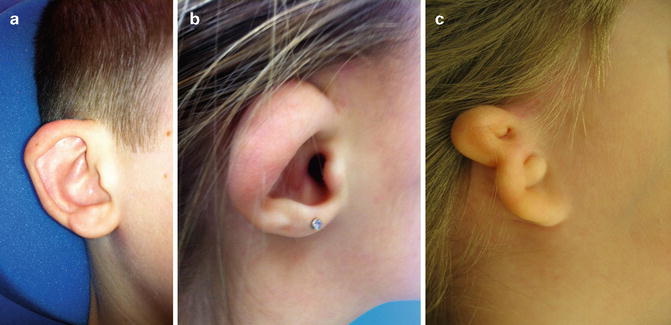
Table 2.6
De la Cruz classification of congenital aural atresia
Minor malformations | Major malformations |
|---|---|
Normal mastoid pneumatization | Poor pneumatization |
Normal oval window/footplate | Abnormal or absent oval window/footplate |
Reasonable facial nerve–footplate relationship | Abnormal course of the facial nerve |
Normal inner ear | Abnormalities of the inner ear |
Table 2.7
Weerda’s combined classification of auricular defects, including surgical recommendations
First-degree dysplasia. Average definition: Most structures of a normal auricle are recognizable (minor deformities) |
Surgical definition: Reconstruction does not require the use of additional skin or cartilage |
a. Microtia |
b. Protruding ear (synonyms: prominent ear, bat ear) |
c. Cryptotia (synonyms: pocket ear, group IV B (Tanzer)) |
d. Absence of upper helix |
e. Small deformities: Absence of the tragus, satyr ear, Darwinian tubercle, additional folds (Stahl ear) |
f. Colobomata (synonyms: clefts, transverse coloboma) |
g. Lobule deformities (pixed lobule, macrolobule, absence of lobule, lobule colobamata (bifid lobule)) |
h. Cup ear deformities |
Type I: Cupped upper portion of the helix, hypertrophic concha, reduced height (synonyms: lidding helix, constricted helix, group IV A (Tanzer), lop ear) |
Type II: More severe lopping of the upper pole of the ear; rib cartilage is used as support when a short ear must be expanded or the auricular cartilage is limp |
Second-degree dysplasia |
Average definition: Some structures of a normal auricle are recognizable |
Surgical definition: Partial reconstruction requires the use of some additional skin and cartilage. Synonym: Second-degree microtia (Marx) |
a. Cup ear deformity, type III: The severe CUP ear deformity is malformed in all dimensions (synonyms: cockleshell ear, constricted helix, group IV (Tanzer), snail-shell ear) |
b. Mini-ear |
Third-degree dysplasia |
Average definition: None of the structures of a normal ear is recognizable |
Surgical definition: Total reconstruction requires the use of skin and large amounts of cartilage. Synonyms: complete hypoplasia group II, peanut ear, third-degree microtia (Marx); normally concomitant congenital atresia is found |
a. Unilateral: One ear is normal; no middle ear reconstruction is performed on any child; auricle reconstruction is begun at age 5 or 6 years |
b. Bilateral: Bone-conduction hearing aid before the first birthday; middle ear surgery at age 4 years without transposition of the vestige; bilateral reconstruction of the auricle at age 5 or 6 years |
Anotia—Complete absence of any recognizable external ear structures. Management is similar to that of third-degree dysplasia |
Fig. 2.6
(a) Photograph of child with first-degree dysplasia of pinna, demonstrating cupped ear, absent antihelical fold, and Darwinian tubercle. (b) Photograph of child with second-degree dysplasia demonstrating underdeveloped upper half of pinna. (c) Photograph of child demonstrating third-degree dysplasia. Note that the normal structures of the ear are not recognizable
Anotia
Complete absence of any recognizable external ear structures. Management is similar to that of third-degree dysplasia.
Diagnosis
Microtia is evident on initial neonatal assessment. A thorough physical examination is important to accurately describe the auricular deformity, determine the status of the EAC, and assess for any associated malformations (preauricular pits/sinuses, cervical cysts, or sinuses), craniofacial malformations (ocular, zygomatic, mandibular, palatal abnormalities), as well as other organ systems. This information will be helpful in determining a possible syndromic etiology. Microtia, as with CAA, is recognized by the American Academy of Pediatrics as a high-risk factor for congenital hearing loss. It is paramount that the hearing status of a neonate with a diagnosis of microtia be promptly determined. With bilateral microtia, early referral for audiologic evaluation is critical so that proper bone-conduction hearing aid use can be implemented. With unilateral microtia, a diagnostic ABR test is typically recommended to ensure that the child has at least one normal hearing ear. If there is associated CAA and surgical correction is contemplated, a preoperative CT scan is recommended.
Management
Management of microtia is commonly a multidisciplinary approach. If other congenital anomalies are present, either in association with a syndrome or as isolated findings, consultation from the appropriate services is indicated. From an otologic perspective, one must initially determine what type of auditory habilitation is required and feasible. Patients with microtia associated with CAA follow the algorithm previously discussed under CAA, as auditory habilitation of the associated CHL takes precedence in these cases. If surgery is contemplated, microtia repair is undertaken at approximately 5–6 years of age, with atresia repair following at approximately 6–7 years.
Using the Weerda classification, first-degree dysplasia encompasses many common and frequently isolated auricular malformations, including macrotia, protruding ears, and cup ear deformity. Reconstruction normally does not require the use of additional skin or cartilage. Instead, many otoplastic techniques have been described that attempt to achieve some auricular reduction. These techniques for the most part involve manipulation of the cartilage framework by using sutures or by cutting, abrading, or scoring. Examples of the former include the Mustarde and the Furnas techniques. Examples of the cartilage-cutting techniques include the Converse, Farrior, and Pitanguy techniques. These techniques make use of the observation that cut cartilage bends away from its cut side. Cartilage cutting is useful for manipulating cartilage that is stiff and thick but can cause scarring, irregularities along the cut edges, and a diminished auricle size. They are also generally more difficult to undertake and require an experienced surgeon for their maximal benefit and utilization.
Second-degree dysplasia, or atypical microtia, has most of the auricular structural components recognizable; there is however distinct tissue deficiency that necessitates the transposition of skin and cartilage. The main deficiency often lies in the vertical height of the affected ear. Augmenting this deficiency can be achieved by various techniques depending on the difference in height between the two ears. If the height difference is more than 20 mm, a cartilage graft is considered. Donor sites include the contralateral conchal bowl or a rib graft for large deficiencies. A staged skin graft in these instances is often required and may be advanced from a postauricular donor site.
Reconstruction for third-degree dysplasia (classic microtia) or anotia requires the use of skin and large amounts of cartilage. This is best initiated at the age of about 6 years, especially for unilateral cases, and the best donor tissue is autogenous costal cartilage. By this age, sufficient cartilage is present for the auricular reconstruction, and the patient is more able to cooperate with the necessary postoperative care. The reconstruction is multistaged and incorporates CAA repair.
Middle Ear
Embryology
The tympanomastoid compartment first appears at the 3-week stage as an outpouching of the first pharyngeal pouch (the tubotympanic recess) (Fig. 2.7). This is a stalklike diverticulum, which expands in a lateral direction until it comes in contact with the epithelial lining of the first pharyngeal cleft. The distal part of this diverticulum widens into a saclike structure, which is the primitive tympanic cavity. Expansion of the pouch begins at the inferior aspect of the definitive tympanic cavity and progresses by invasion of the adjacent mesenchyme. By 7 weeks, concomitant growth of the second branchial arch constricts the importation of the tubotympanic recess. The primary tympanic cavity lies lateral to this constriction, while the proximal part of the tubotympanic recess medial to this constriction remains narrow and develops into the Eustachian tube. The distal end of the first pharyngeal pouch buds into four sacci (anticus, posticus, superior, and medius), which expand to progressively pneumatize the middle ear and the epitympanum. Expansion of the sacci envelops the developing ossicles, which remain embedded in mesenchyme until the eighth month, when the surrounding tissue dissolves. The interface between two sacci gives rise to mesentery-like mucosal folds, which transmit blood vessels. Mesenchymal resolution may continue as late as 1 year postnatally (or even later in some rare cases). Persistence of this embryonic connective tissue in the adult may be evident as connective tissue strands draped over the oval and round windows.
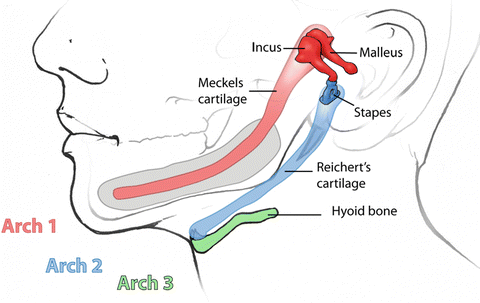
Fig. 2.7
Schematic diagram demonstrating the origin of ossicles and their relationship to the branchial arches
By birth, the antrum approximates that of the adult. Mastoid pneumatization is evident as early as 33 weeks gestation and proceeds along well-established tracts. There is considerable variability amongst individuals in the degree and pattern of temporal bone pneumatization, and multiple factors are thought to play a role in this, including heredity, environmental, nutritional, infectious, and anatomic factors relating to the adequacy of ventilation by the Eustachian tube. Overall, the mastoid continues to grow for up to 19 years after birth.
The ossicular chain largely has its origins traced back to the pharyngeal arch apparatus, and studies have established that it is the phylogenetic equivalent of the jaw joint in all nonmammalian jawed vertebrates [56]. In these animals, the jaw joint is established between mesenchymal components of the first and second pharyngeal arches, namely, the quadrate and articular portions. In the typical living reptile, sound is transmitted through the columella auris from a primitive tympanic membrane to the inner ear [57]. During evolution, the jaw joint was modified, and the articulo-quadrate joint evolved to constitute the mammalian middle ear [58]. The articular became the malleus and the quadrate the incus. These ossicles, through their respective contacts with the tympanic membrane and stapes (the phylogenetic homolog of the columella auris), were incorporated into the sound-transmitting apparatus [59, 60].
The first evidence of ossicular development occurs at approximately 4 weeks. An interbranchial mesenchymal bridge appears between the first and second pharyngeal arches. Specifically, the bridge connects the upper end of a part of the first arch (referred to as the mandibular visceral bar) with the central region of the second pharyngeal arch (hyoid) visceral bar. Thus, this condensed mesenchymal bridge consists of both first and second pharyngeal arch elements, which ultimately undergoes cartilaginous differentiation that gives rise to the primordial malleus and incus. As for the stapes, all of its blastula derives from the hyoid visceral bar except for the medial surface of the footplate and the annular ligament, which are of otic capsular (lamina stapedialis) origin. Thus, the ossicles are derived from neural crest mesenchyme of the first and second branchial arches, with the only exception being the medial aspect of the stapes footplate and the annular ligament, which are derived from the mesenchyme of the otic capsule.
Over the following 11 weeks, the ossicles develop and grow as a cartilaginous model of the mandibular and hyoid arches. This process is bone formation from a cartilaginous framework and is referred to as endochondral ossification. The anterior process of the malleus is unique in that it develops by membranous ossification without a cartilaginous model. Although controversy exists as to the exact contribution of each arch to the ossicular chain, it is believed that the head of the malleus as well as the body and short process of the incus are formed from Meckel’s cartilage. Meanwhile, the long process of the incus, handle of the malleus, stapes superstructure, and tympanic surface of the stapes footplate are derived from Reichert’s cartilage. As previously mentioned, the medial surface of the footplate and the annular ligament are of otic capsular origin. Development of the stapes blastema involves progressive encirclement of the stapedial artery. The obturator foramen represents the completed ring left empty after the stapedial artery involutes.
By 15 weeks of gestation, the ossicles have already attained adult size, and the process of endochondral ossification soon begins. This process starts first in the incus, then in the malleus, and finally in the stapes. As the footplate attains adult size, the annular ligament is formed from the developing otic capsule. Paralleling this development, the tensor tympani and stapedius muscles develop. The tensor tympani muscle is innervated by the mandibular branch of the trigeminal nerve, the nerve of the first pharyngeal arch, while the stapedius muscle is innervated by the facial nerve, the nerve of the second pharyngeal arch. By 20 weeks gestation, the ossicles have assumed their adult configuration, and the endochondral bone of the ossicles, as with that of the otic capsule, undergoes little change over the lifetime of the individual. The exception to that is the stapes, which continues to lose bulk from its large structure well into the 32nd week of gestation. The stability of the ossicular bone explains the poor reparative capacity in response to trauma.
Although the ossicles appear during the first half of fetal life, they remain embedded in mesenchyme until the eighth month, when the surrounding tissue dissolves. As the mesenchyme surrounding the ossicles dissolves and the developing tympanic cavity expands, the endodermal epithelial lining of the primitive tympanic cavity then extends along the wall of the newly developing space to the epitympanic space and antrum and envelopes the developing ossicles. The tympanic cavity is now at least twice as large as before. When the ossicles are entirely free of surrounding mesenchyme, the endodermal epithelium connects them in a mesentery-like fashion to the wall of the cavity. The supporting ligaments of the ossicles develop later within these mesenteries.
During late fetal life, the tympanic cavity expands dorsally by vacuolization of surrounding tissue to form the tympanic antrum. After birth, epithelium of the tympanic cavity invades bone of the developing mastoid process, and epithelium-lined air sacs are formed (pneumatization). Later, most of the mastoid air sacs come in contact with the antrum and tympanic cavity.
Ossicular Chain Malformations
Definition
Ossicular chain malformations describe a spectrum of anomalies that may include absence or maldevelopment of any of the ossicles (Fig. 2.8).
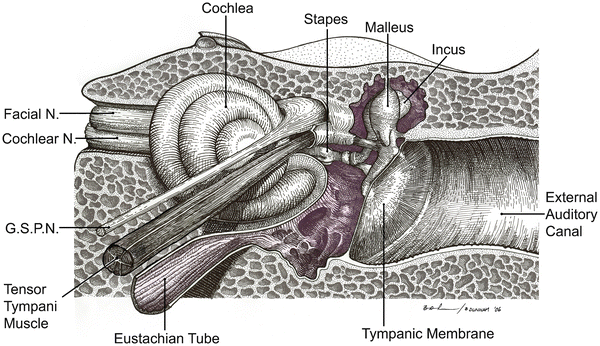
Fig. 2.8
Schematic of normal anatomy of middle ear
Incidence
Overall, isolated congenital ossicular anomalies are rare, with an incidence of less than 1 per 15,000 births [61]. In a study of 565 children with congenital hearing loss, 54 children (9.5 %) had CHL unrelated to otitis media and only 3 children (0.5 %) were found to have an isolated middle ear anomaly [62]. In a similar study of 687 children with congenital hearing loss, 8 children (0.1 %) had an isolated middle ear defect [42]. The most common congenital isolated ossicular anomalies in many series remain stapes fixation and incudostapedial discontinuity [63, 64], with isolated congenital stapes fixation representing 20–50 % of ossicular malformations [64–67].
Etiology
Etiologic factors related to ossicular chain abnormalities remain unclear. It would appear that most of these isolated anomalies arise from the lower part of the ossicular chain and are related to the second pharyngeal arch. It is yet unclear as to why anomalies are often restricted only to the stapes, or why other structures derived from the second arch are not affected with these ossicular anomalies. Congenital X-linked mixed deafness is a rare anomaly that occurs in males and is typified by progressive mixed hearing impairment from stapes fixation with perilymphatic gusher. It is inherited in an X-linked recessive pattern, and male patients tend to have severe mixed hearing loss at all frequencies, while female carriers have normal hearing or mild hearing loss. The condition is associated with anomalies of the vestibule and internal auditory canal (IAC). The gene defect has been localized to the Xq13–q21.1 region, which encodes the POU3F4 transcription factor [68]. The etiology of epitympanic fixation of the head of the malleus is rooted in the incomplete pneumatization of the epitympanum.
Associated Malformations
Because of the shared embryologic derivative, external ear abnormalities are often associated with middle ear abnormalities, and CAA can be associated not only with auricular but also with middle ear abnormalities. However, it is important to remember that the auricle, EAC, and ossicular chain originate from different segments of neural crest cells of the first and second pharyngeal arches and develop at different times in embryonic development. As such, a normally developed ossicular chain may be present despite a severe microtia with accompanying CAA.
Ossicular abnormalities may be associated with altered anatomy of middle ear structures. Jahrsdoerfer found the incidence of having an aberrant facial nerve course in ears with a congenital middle ear malformation was 24 % [69]. Ossicular chain abnormalities may also be seen in association with craniofacial anomalies. Many reports have found associations with congenital syndromes in approximately 20 % of middle ear malformations [70, 71]. The more common craniofacial syndromes involving conductive hearing losses are Treacher Collins, Crouzon, Apert, Goldenhar, BOR, Pfeiffer, Beckwith–Wiedemann and Klippel–Feil syndrome, as well as, CHARGE Association (coloboma, heart defects, atresia of the choanae, retarded growth or development of the central nervous system, genitourinary anomalies, and ear anomalies) syndromes.
Clinical Features
Congenital stapes fixation (SF) is often encountered in minor malformation cases and usually presents as a CHL with a patent EAC and normal TM. The differential diagnosis of stapes fixation includes other ossicular malformations, oval or round window atresia, congenital cholesteatoma, middle ear tumors including vascular lesions, and ossicular trauma. SF is bilateral in 70–90 % of patients [67, 71]. SF should be distinguished from juvenile stapes otosclerosis, which is defined by a progressive CHL. Furthermore, congenital SF presents at an earlier age than does juvenile otosclerosis (age 3 years vs. 10 years) [72]. Half of the children with juvenile otosclerosis have a positive family history; only 10 % of those children with congenital SF have other family members with CHL.
Classification
Classification of ossicular abnormalities can be divided into major and minor. Minor congenital ossicular anomalies are restricted to the middle ear, while major congenital ossicular anomalies can involve the auricle, EAC, and middle ear cleft. Different classifications are based on anatomic findings and/or embryologic malformations; however, none are universally accepted. Cremers’ [73] and Charachon’s [74] classifications are amongst the most commonly used (Tables 2.8 and 2.9).
Table 2.8
Cremers’ classification of congenital middle ear anomalies
Class 1 | Congenital stapes ankylosis without other deformities in the middle ear |
Class 2 | Congenital stapes ankylosis in combination with a congenital anomaly of the ossicular chain |
Class 3 | Congenital anomaly of the ossicular chain, but mobile footplate |
3a | Discontinuity of the ossicular chain |
3b | Epitympanic fixation |
Class 4 | Congenital aplasia or severe dysplasia of the oval or the round window |
Table 2.9
Charachon’s classification of congenital middle ear anomalies
Class 0 | Normal ossicular chain, almost normal tympanic membrane but small atretic plate around malleus handle |
Class 1 | Fixation of the malleus head |
Class 2 | Normal ossicular chain but fixation of the footplate |
2a | With abnormality of the facial nerve |
2b | Without abnormality of the facial nerve |
Class 3 | Lack of a part of the ossicular chain with or without abnormality of the stapes |
Class 4 | Severe malformation of all the ossicular chain |
Diagnosis
The diagnosis is suspected in a patient who presents with stable hearing loss present since birth without a history of recurrent ear infections or trauma. Otomicroscopy is often normal, but it may reveal an abnormality of the TM, malleus, or incus. An audiogram, in most cases, may demonstrate CHL in the 40–60 dB range. Impedance testing may be useful in the evaluation of some of these pathologies, as in congenital footplate fixation where typically there is absence of acoustic reflexes and a type A-shallow tympanogram. The hearing loss does not worsen with time, unless there is an associated Eustachian tube dysfunction, which is often seen in syndromic cases, and can lead to diagnostic difficulties. Generally, the diagnosis is easier and made earlier when the malformation is bilateral; in unilateral cases, a diagnosis is typically established at the age of 6 years. High-resolution CT scan may reveal the presence of various ossicular anomalies. Surgery in the form of exploratory tympanotomy offers a means for definitive diagnosis and the option of surgical correction at the same time.
Management
It is reasonable to offer hearing aids and defer surgical exploration until the child is older, when a series of hearing tests are available, and the child is more able to share in the consent-making process. A CT scan should be obtained at some point during the child’s follow-up period. Imaging is useful in that it can provide essential diagnostic information to elucidate the cause of the CHL and identify associated middle or inner ear abnormalities. Concerning inner ear findings include an enlarged vestibular aqueduct, dilated IAC fundus, and an abnormal communication between the inner ear and intracranial space.
Given the nonprogressive nature of the hearing loss, amplification is a reasonable option in managing patients with isolated congenital ossicular anomalies. If surgery is contemplated, a comprehensive discussion with the family is essential and waiting until the child is capable of weighing the associated surgical risks, including sensorineural hearing loss, against the potential benefits of surgery. The rate of severe SNHL in patients with fixed stapes has been reported to be between 0 and 30 % in the literature [65, 75, 76]. With regard to patient age, many studies have found no correlation between the patient’s age at the time of stapedectomy for congenital SF and postoperative audiometric results [77]. Many studies have reported good results following stapedectomies for congenital SF [67, 71, 74, 78]; however, hearing outcomes tend to be worse compared with the results for otosclerosis [79–81]. This may be attributable to the higher incidence of coexisting middle ear anomalies with congenital SF. The reported success rate of ossicular chain reconstruction is usually lower than that of stapedectomy and varies widely between series. Ossiculoplasty for combined ossicular anomalies yields an air–bone gap (ABG) of less than 30 dB in about 70 % of patients.
Congenital Cholesteatoma
Definition
Cholesteatoma refers to a collection of keratinizing squamous epithelium that can be trapped within the temporal bone and grows into a destructive lesion. Left untreated, a cholesteatoma can be a significant source of morbidity as it eventually erodes into critical structures, including the ossicles, facial nerve canal, inner ear, and tegmen. The latter may result in severe intracranial complications and even death. Cholesteatomas may be congenital or acquired. In the past, the diagnosis of congenital cholesteatoma (CC) depended only on the presence of a white mass behind an intact tympanic membrane in a child with no previous history of otitis media. The mass is often found within the anterior-superior portion of the middle ear. However, due to the high incidence of acute otitis media (60–80 % by 1 year of age, and 80–90 % by 2–3 years) [82, 83], the criteria have been modified to define a CC as a whitish mass behind an intact TM, with the absence of otorrhea or perforation and no previous otologic procedures, including myringotomy or insertion of ventilation tubes. Studies have since shown that the diagnosis should also not be excluded in cases of otorrhea. or myringotomy [84].
Incidence
CC accounts for approximately 4 % of childhood cholesteatoma [84]. The number of reported cases of CC has increased dramatically over the last three decades, and this may be related to a better understanding and awareness of the disease entity, leading to timely diagnosis as well as improved otomicroscopic diagnostic tools. Improved medical management of otitis media may have led to a decrease in the number of acquired cholesteatomas and a relative increase in the percentage of the congenital type.
Etiology
The most popular theory of CC formation states that squamous inclusion cysts arise from epithelial rests (epidermoid formations) in the middle ear. These epidermoid formations have been demonstrated histologically in fetal temporal bones, and they may be single or multiple. They typically disappear in the third trimester of gestation. Failed involution leads to cholesteatoma formation behind an intact TM. Alternative but less supported theories include seeding of the middle ear by squamous cells in the amniotic fluid or from the surface epithelium of the TM after infection and microperforation.
Associated Malformations
CC has been associated with congenital ossicular malformations, including defects of the long process of the incus and the stapes superstructure. CC has also been associated with cholesterol granuloma and abnormal vestibular anatomy (dilated endolymphatic fossa, large vestibular aqueduct, and hypoplastic vestibule).
Clinical Features
The average age of presentation of children with CC is 3–5 years [84, 85]. The chief presenting symptom varies depending on the stage of the cholesteatoma. Asymptomatic cases are most prevalent. In symptomatic cases, hearing loss is the most prevalent symptom. Less common presenting symptoms include aural fullness, otalgia, tinnitus, dizziness, or headache. Facial nerve paralysis is rare and constitutes less than 1 % [84, 86]. CC may be found incidentally at myringotomy for serous otitis media.
Classification
Potsic et al. [84] proposed staging system for CC based on preoperative CT scans (Table 2.10). This system takes into consideration middle ear and mastoid extension as well as ossicular involvement. Nelson et al. [87] proposed a three-stage classification system based on the presumed natural history of these lesions, tracking them as they start in the anterior-superior quadrant, then extend to the anterior-inferior quadrant before progressing into the posterior-superior quadrant, the attic, and finally into the mastoid (Table 2.11). In their series, this classification system correlated with the degree of CHL and the risk of recurrence as well as escalating complexity of surgical approach.
Table 2.10
Potsic staging system for congenital cholesteatoma
Stage I | Single quadrant: No ossicular involvement or mastoid extension |
Stage II | Multiple quadrants: No ossicular involvement or mastoid extension |
Stage III | Ossicular involvement: Includes erosion of ossicles and surgical removal for eradication of disease; no mastoid extension |
Stage IV | Mastoid extension (regardless of findings elsewhere) |
Table 2.11
Nelson classification system for congenital cholesteatoma
Type I | Confined to the middle ear but does not involve the ossicles, except for the manubrium |
Type II | Involvement of the ossicular mass in the posterior superior quadrant and the attic |
Type III | Mastoid extension |
Diagnosis
CC may be diagnosed on otomicroscopy of an asymptomatic child or one being evaluated for hearing or other otologic symptoms. Early on, the cyst is hard to appreciate, appearing as a subtle whitish discoloration behind an otherwise normal TM (Fig. 2.9a). As the lesion grows, it becomes more evident as it contacts the medial aspect of the TM, which it may displace laterally. If the lesion obstructs the Eustachian tube (ET), it may result in a middle ear effusion, and this may complicate the diagnosis. With further progression, the cyst may impinge or erode the ossicles, worsening the conductive hearing loss. A thorough history should detail previous otologic procedures, ear infections, or temporal bone trauma. Complete audiometric assessment assesses for the presence and degree of CHL, and a preoperative CT scan is used to delineate disease extent, assess for temporal bone anatomy that may be used as a roadmap during surgery, and identify any alteration including scutum erosion and facial nerve dehiscence (Fig. 2.9b and c).
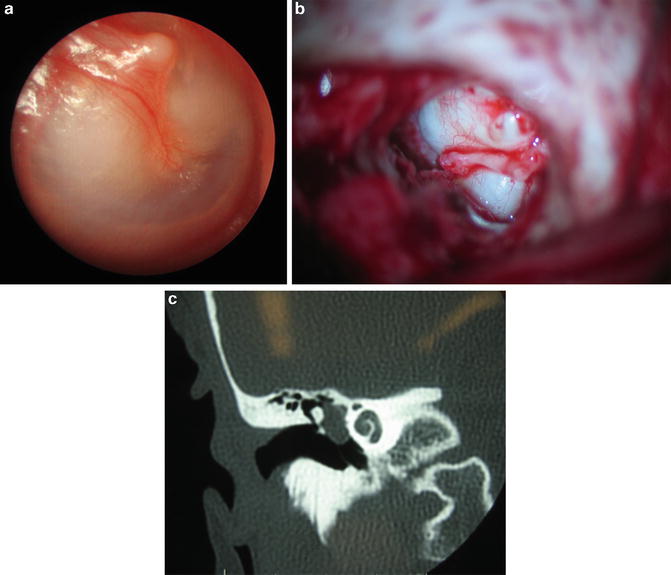
Fig. 2.9




(a) Photograph of child with congenital cholesteatoma behind an intact, bulging eardrum. (b) Intraoperative photograph of middle ear with elevated eardrum demonstrating ossicles and cholesteatoma. (c) CT scan demonstrating opacity in middle ear (congenital cholesteatoma) and intact ossicles (all images courtesy of John A. Germiller, MD, PhD)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
