Fig. 3.1
Embryology of the development of the nose
The deepening of the nasal pits leads to the formation of the primordial nasal sacs, which grow dorsally. The oronasal membrane, which first separates the nasal sacs from the oral cavity, ruptures by the end of the sixth week forming an open communication between the nasal and oral cavities. A temporary epithelial plug is formed in the nasal cavity from the expansion of cells lining it; this plug resorbs between the 13th and 15th weeks. The primordial choanae lie posterior to the primary palate. After the secondary palate develops, the choanae position themselves at the junction of the nasal cavity and the pharynx. As the maxillary palatal shelves lengthen and grow medially, they fuse with each other and the septum, effectively separating the nasal cavities from the oral cavity [4]. Eruption of the nasal pits into the choanae, fusion of the palatal shelves and growth of the nasal septum and soft palate coincide with the development of the lateral nasal wall and primitive sinus anatomy.
Congenitally Malformed Nose
Arhinia
Definition
Arhinia is defined as the congenital absence of a nose (Fig. 3.2).

Fig. 3.2
Diagram of a child with arhinia
Incidence
Arhinia is an extremely rare condition. All cases are thought to be sporadic.
Etiology
The pathogenesis of arhinia is poorly understood. It has been postulated that lack of development of the nose results from failure of the medial and lateral nasal processes to grow, but it is also possible that overgrowth and premature fusion of the nasal medial processes result in formation of an atretic plate. Arhinia may also result from lack of resorption of the nasal epithelial plugs during the 13th–15th weeks of gestation. Another explanation may be related to abnormal migration of neural crest cells to this region, resulting in aberrant flow of the multiple mesodermal structures required to establish the nose and its cavities normally [5].
Associated Malformations
Microphthalmia, iris coloboma, hypertelorism, submucous cleft palate, and meningocele occasionally associate with this condition. Intelligence is usually normal.
Clinical Features
Neonates are obligate nasal breathers and become symptomatic with respiratory distress and cyanosis when the nasal passages are blocked, especially during feeding. Affected infants, however, can survive and adapt to oral breathing. The site of the external nose is flat, covered with normal appearing skin. Occasionally, a blind dimple is present in lieu of the nostrils. While the upper lip is normal, the palate is high-arched and the maxilla hypoplastic. The nasal cavity fails to form and there is bilateral bony choanal atresia [2].
Diagnosis
Diagnosis is based on visual inspection. Computed tomography imaging of the nose and midface is recommended to evaluate the underlying structure. Evaluation of the thickness of the atretic bony plate is an important preoperative consideration.
Management
An oral airway is often used in the neonatal setting. A surgically created nasal airway or a tracheostomy tube is an important part of early management, as either allows the infant to feed orally and precludes the complications associated with orogastric tubes. Most authors agree that surgical reconstruction of the external nose and inner cavities should be delayed at least until preschool years, when facial development is nearly complete [6, 7].
Unilateral Arhinia, Heminasal Aplasia
Definition
Unilateral arhinia is the unilateral absence of one nostril. Occasionally, a blind dimple is present in lieu of a nostril. It differs from cebocephaly, which presents as a single midline nostril. Cebocephaly is a congenital condition with monkey-like facial features with closely set eyes, a defective or absent nose, and a tendency towards cyclopedia (Fig. 3.3).
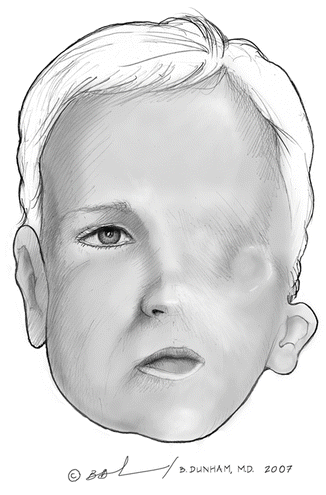
Fig. 3.3
Diagram of a child with unilateral arhinia
Incidence
Unilateral arhinia is extremely rare and is sporadic.
Associated Malformations
The condition may be associated with a blind dimple, skin tag, or a proboscis [8]. There are a significant number of bony abnormalities associated including an absent cribriform plate, nasal septal deviation towards the affected side, malformation of the ipsilateral lateral nasal wall, absence of the nasal bones, and disruption of the lacrimal bone. The ipsilateral olfactory tract and bulb are typically absent. The ipsilateral eye may be microphthalmic, absent, or anomalous. Orofacial clefting has been seen in some cases. In the absence of associated central nervous malformations, intelligence is usually normal.
Diagnosis
The diagnosis is based on visual inspection and computed tomography of the midface.
Nostril Coloboma
Definition
A nostril coloboma is a triangular cleft of the nasal ala (Fig. 3.4).
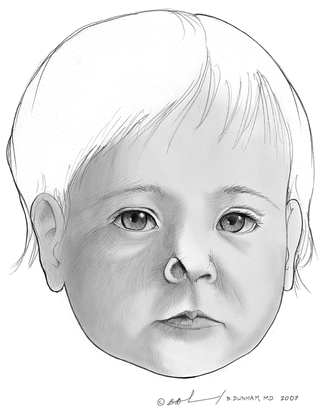
Fig. 3.4
Diagram of a child with a nostril colomba
Incidence
Unilateral nostril coloboma is rare and sporadic. Bilateral nasal colobomas occur even less frequently.
Associated Malformations
Unilateral anophthalmia, lower lid coloboma, frontal glioma, and ocular hypertelorism can occur simultaneously.
Diagnosis
Its diagnosis is made by visual inspection. The defect is typically unilateral.
Bifid Nose
Definition
In this rare and sporadic condition, a central groove bifurcates the nose. The condition may result from an anomalous infolding of the median nasal processes (Fig. 3.5).
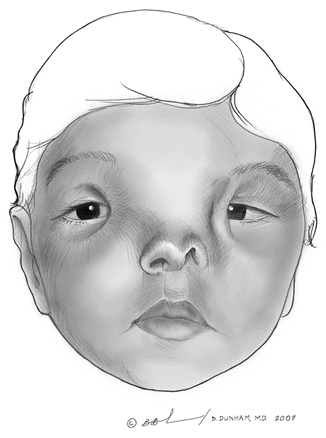
Fig. 3.5
Diagram of a child with a bifid nose
Incidence
The incidence of these rare clefts has been estimated at 1.43–4.85 per 100,000 births [9].
Associated Malformations
Ocular hypertelorism often occurs concurrently. The upper lip may also have a median cleft. Pseudohypertelorism can also occur as a result of an optical illusion. This illusion is produced by the wide spacing of the components of the face adjacent to the eyes.
Diagnosis
Visual inspection is the basis for diagnosis. The presentation of a bifid nose ranges from a minimally noticeable midline nasal tip central groove to a complete clefting of the osteocartilaginous framework, resulting in two complete half noses [10].
Management
Surgical repair is the treatment of choice. The rarity of these types of cleft makes any surgeon relatively inexperienced. Not surprisingly, as with many of these malformations, no true consensus exists as to the best approach for repair [9].
Polyrhinia
Definition
Polyrhinia is characterized by either a partial or a complete duplication of the nose (Fig. 3.6).
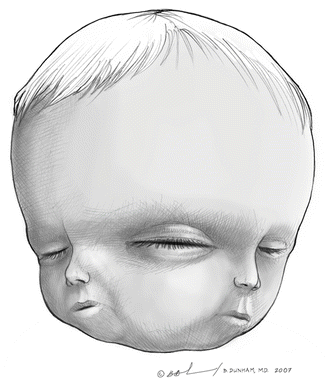
Fig. 3.6
Diagram of a child with polyrhinia and partial facial duplication
Incidence
This is an extremely rare and sporadic condition.
Associated Malformations
Choanal atresia is commonly found in association with polyrhinia. Two separate noses each with a pair of nostrils can occur with diprosopia, a condition of partial facial duplication. Some cases of facial duplication are incompatible with life.
Diagnosis
Clinical inspection provides the diagnosis.
Management
If the child does survive, however, treatment is surgical [2]. In the absence of facial duplication, treatment involves correcting the associated choanal atresia and then removing the medial portions of both noses and anastomosing their lateral counterparts in the midline.
Proboscis
Definition and Associated Malformations
A proboscis is a snout-like tubular structure arising from the midface (Fig. 3.7). There are four different types: lateral nasal proboscis, supernumerary proboscis, disruptive proboscis, and holoprosencephaly proboscis.
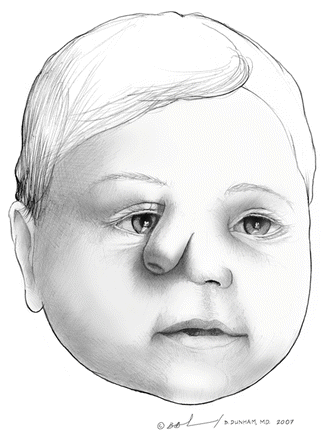
Fig. 3.7
Diagram of a child with proboscis
Lateral nasal proboscis results from the incomplete formation of one side of the nose, including an ipsilateral absence of the nostril, nasal cavity, paranasal sinuses, and olfactory tract and bulb. Typically the proboscis emanates from the medial canthus on the affected side. A supernumerary proboscis occurs in the setting of two nostrils as an accessory structure and is thought to arise from a supernumerary nasal placode. Disruptive proboscis occurs in the setting of an early embryonic hamartoneoplastic lesion in the primitive prosencephalon.
Holoprosencephaly is a failure of the forebrain to divide into lobes; cyclopia occurs in its most severe form. Proboscises occur in many but not all cases of cyclopia. Ethmocephaly, also caused by holoprosencephaly, consists of a proboscis in lieu of a nose in between narrowly set microphthalmic eyes. Prognosis depends on the associated anomalies. Holoprosencephaly forecasts a poor prognosis [2].
Incidence
Proboscis is a rare anomaly. The incidence of proboscis lateralis is less than 1 in 100,000 [11].
Etiology
Proboscis represents a failure of the lateral nasal process to fuse with both the medial nasal process and the maxillary process. The etiology and pathogenesis of proboscises is heterogeneous.
Diagnosis
Clinical inspection provides the diagnosis.
Management
Isolated proboscis formation is amenable to surgical correction. Preoperative imaging with both computed tomography (CT) and magnetic resonance (MR) scans is imperative. Given the high degree of variability of associated anomalies, an individualized approach is suggested when addressing the surgical correction of a proboscis. In general surgical repair can be undertaken as early as the surgeon is comfortable without affecting the cosmetic outcome [12, 13].
Congenital Midline Nasal and Nasopharyngeal Malformations
Choanal Atresia
Definition
Choanal atresia is an uncommon congenital obstruction of one or both of the posterior choanae. The choanae are the spaces that separate the most posterior aspect of the nose from the nasopharynx. About 30 % are purely bony while the remaining 70 % are thought to have mixed bony and membranous atresias [14]. Choanal stenosis may be considered a milder variation of atresia.
History
Roederer was first to describe the condition of congenital choanal atresia in 1755 and Emmert reported the first successful surgical dilation using curved trocars transnasally in 1854 [15].
Incidence
In a review of over five million births, the prevalence at birth varied between 0.54 and 1.13 per 10,000 with an equal sex distribution and without evidence of side predilection [16]. The frequency of unilateral versus bilateral, while it had traditionally been thought to be 2:1, has more recently shown to be closer to 1:1 [17].
Etiology
During development, the nasal cavities extend posteriorly as the palatal processes fuse to form a single closed palate. Failure of the posterior rupture of the buccopharyngeal membrane that separates the nose from the nasopharynx or abnormal growth of the palatine bone may be responsible for choanal atresia. The bony narrowing can result from narrowing of the pterygoid plates laterally, the vomer medially or the sphenoid superiorly.
Associated Malformations
Forty-seven percent of cases have been reported to be associated with other major congenital malformations and should alert physicians to look for other anomalies. Some syndromes associated with choanal atresia include Apert syndrome, Crouzon syndrome, DiGeorge sequence, Pfeiffer syndrome, Treacher-Collins syndrome, and CHARGE association [16]. Choanal atresia is a common finding in CHARGE Association, which is a nonrandom association of malformations whose acronym stands for coloboma, heart defects, and atresia of the choanae, retarded growth or development of the central nervous system (CNS), genitourinary anomalies, and ear anomalies. A child has to have three or more of the cardinal malformations (excluding growth/mental retardation) to meet diagnostic criteria. By those criteria, approximately 7 % of children with choanal atresia belong to the CHARGE constellation [16].
Clinical Features
Bilateral choanal atresia presents very differently than its unilateral counterpart. Because an infant is typically an obligate nasal breather, bilateral obstruction often presents with immediate respiratory distress during the newborn period. The resulting distress is typically cyclical. During the inspiratory effort, the infant’s tongue apposes the palate, occluding the oral airway. Increased inspiratory effort leads to marked retractions. The occlusion is broken if and when the child cries or opens his/her mouth. Asphyxia can and does occur with bilateral atresia.
Diagnosis
The suspected diagnosis is clinically supported by the inability to pass a 6 French suction catheter through the nasal cavity and can be confirmed with a diagnostic flexible nasopharyngoscopy. Computed tomography of the nose can also confirm the diagnosis; furthermore it shows what type of atresia it is, purely bony or membranous and bony (Fig. 3.8). Unilateral atresia rarely causes respiratory distress and often escapes detection until later in childhood when a thick unilateral mucoid discharge is noted; it is not uncommon for it to be diagnosed as sinusitis.
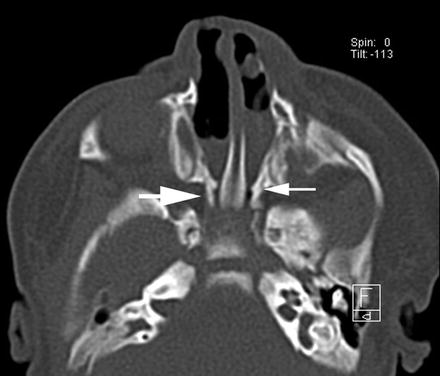
Fig. 3.8
Computed tomography demonstrating choanal atresia (see arrows)
Management
Treatment of choanal atresia is surgical. In the case of bilateral atresia, the immediate distress can often be temporarily addressed with either an oral airway or a McGovern nipple, which has either a single enlarged hole or two additional lateral holes at its tip. There are three basics approaches for the repair: transpalatal, transeptal, and transnasal. The transpalatal approach, while it provides great visualization of the surgical field, is typically reserved for older patients as it can result in malocclusion in up to 50 % of patients, a consequence of disrupting the palate before it has completely grown [18]. The transeptal approach is typically reserved for unilateral cases. Endoscopic transnasal repair is the most common approach today; it can address either membranous or bony defects. Endoscopes can be used transnasally and through the oropharynx (Fig. 3.9) demonstrates the endoscopic appearance of the bilateral atresia taken from the nasopharynx. Revision surgery is commonly needed to dilate the passages as the child grows.
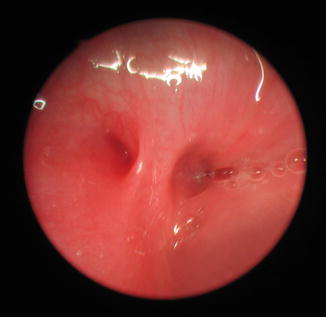
Fig. 3.9
Endoscopic photograph of bilateral choanal atresia taken from the nasopharynx
Craniopharyngioma
Definition
Craniopharyngiomas are benign-appearing dysodontogenic epithelial tumors; the great majority of these affect intradural suprasellar anatomy but they can and do extend into infrasellar regions.
Incidence
The overall incidence of craniopharyngiomas is approximately 0.13 per 100,000 person years and is not gender or race dependent. Craniopharyngiomas comprise approximately 1.5–11.6 % of all intracranial tumors. A bimodal distribution places peak incidence rates in children (aged 5–14 years) and adults (aged 50–74). Approximately 338 cases of this disease occur annually in the United States, with 96 occurring in children from 0 to 14 years of age [19].
Etiology/Embryology
Craniopharyngiomas are thought to arise from the remnants of Rathke’s pouch, which arises during the fourth week of gestation from the oral stomodeum and projects dorsally towards the brain, eventually forming the anterior lobe and pars intermedia of the pituitary gland (Fig. 3.10). By the eighth week, Rathke’s pouch has typically lost its contact with the stomodeum [4]. By the 12th week, the craniopharyngeal duct, the cellular tract formed by the dorsal ascension of Rathke’s pouch disappears, leaving an obliterated tract between the sphenoid cavity to the junction of the palate and posterior nasal septum. Most craniopharyngiomas occur in and/or above the sella turcica. Only occasionally do they develop in the basisphenoid or pharynx, presumably from rest of cells in an incompletely obliterated tract.
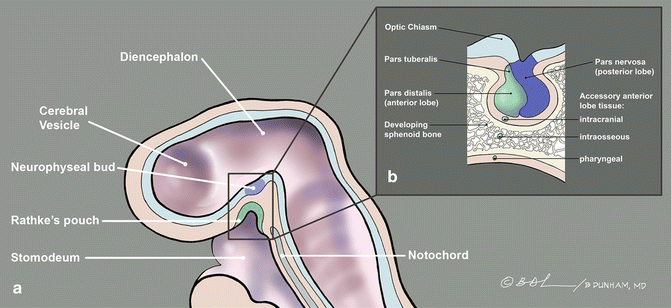
Fig. 3.10
Schematic diagram demonstrating the possible mechanism of formation of a craniopharyngioma
Associated Malformations
If there is a suprasellar/intracranial component, then visual field defects, varying degrees of pituitary insufficiency and signs of increasing intracranial pressure may appear.
Clinical Features
Craniopharyngiomas’ symptomatology results from local expansion and impingement on surrounding structures; metastases from craniopharyngiomas are extremely rare. In the case of a purely infrasellar mass, nasal obstruction, epistaxis, sinusitis, and headaches predominate.
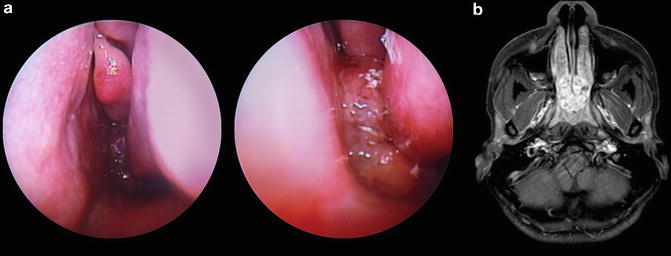
Diagnosis
Initial suspicion of an intranasal mass is made either by nasal endoscopy or imaging (Fig. 3.11a, b). The final diagnosis relies on histopathology. The work up should include general and neurologic examination (including visual field evaluation), a thorough nasal endoscopy, and both magnetic resonance imaging with and without angiography (MRI/MRA) and CT imaging with and without contrast of the brain and sinuses. CT imaging will often detect the calcifications found in the adamantinomatous variant as well as bony destruction and remodeling. MRI imaging best delineates soft tissue involvement and is particularly important for preoperative planning. If intracranial involvement is noted, an endocrine workup is indicated.