Fig. 6.1
Embryology of the larynx
The primitive larynx is first evident at 32 days of gestation when the mesenchymal arytenoid swellings derived from the sixth branchial arch form on either side of the laryngeal orifice and abut the tissue superiorly (hypobranchial eminence), creating a T-shaped aditus. The hypobranchial eminence gives rise to the epiglottis, cuneiform cartilages, and supraglottic structures. The arytenoid swellings fuse in the midline, and then grow cranially to differentiate into the arytenoid and corniculate cartilages and aryepiglottic folds. The thyroid cartilage develops from surrounding tissue from the fourth branchial arch. During this time, the laryngeal epithelium proliferates causing a temporary occlusion of the laryngeal lumen, which then recanalizes allowing the glottis to be patent and for the laryngeal ventricles to form. Failure of recanalization may lead to webs or atresia of the supraglottis, glottis, or subglottis (refer to below).
Endoscopic Anatomy of the Larynx
The larynx consists of a cartilaginous skeleton, intrinsic and extrinsic muscles, and mucosal lining. The larynx can be divided into three sections: the supraglottis (including superior to the ventrical, the epiglottis, aryepiglottic folds, and false cords), the glottis (the vocal folds), and the subglottis. The saccule is a small diverticulum lined by mucous glands extending upward from the anterior ventricle of the larynx and the inside surface of the thyroid cartilage lamina. The piriform sinus is located within the hypopharynx and is lateral to the laryngeal orifice, bounded medially by the aryepiglottic fold and laterally by the thyroid cartilage and hypothyroid membrane. The vallecula is a space bounded by the lingual surface of the epiglottis and base of the tongue. The postcricoid space is posterior to the arytenoid cartilages (Fig. 6.2). Figure 6.3a, b depict the normal larynx and vocal cords as viewed during direct laryngoscopy.
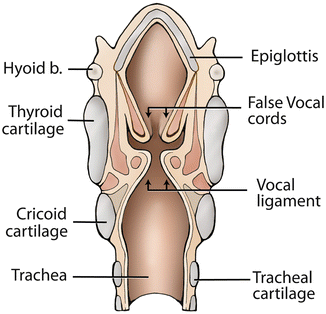
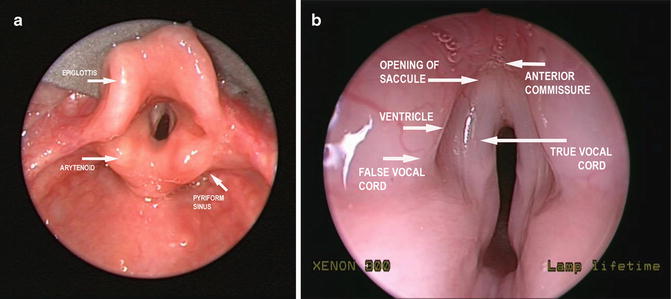
Fig. 6.2
Illustration demonstrating the anatomy of the larynx
Fig. 6.3
Endoscopic photographs demonstrating the surface anatomy of a normal larynx
Laryngomalacia
Definition
Laryngomalacia is a condition in which laryngeal tone is weak, resulting in dynamic prolapse of supraglottic tissue into the airway [1]. This condition is characterized by inspiratory stridor and is associated with varying degrees of airway obstruction.
History
Laryngomalacia was first described by Jackson and Jackson in 1942 in relation to a flaccid larynx and the collapse of supraglottic tissue onto the glottis during inspiration [2]. Historically, severe cases of laryngomalacia required placement of a tracheotomy. In the 1970s, supraglottoplasty was introduced. This currently remains the operative procedure of choice [3].
Incidence
Heredity
The mode of inheritance has not been defined.
Etiology
Although there is no consensus as to the etiology of laryngomalacia, several theories have been proposed. The anatomic theory suggests that characteristic stridor and airway obstruction are caused by the abnormal anatomic location of flaccid laryngeal tissue. The cartilaginous theory proposes that the disease is caused by immaturity and pliability of the laryngeal cartilages. The neurologic theory includes multiple mechanisms including neuromuscular hypotonia, disrupted cortical function, and abnormal neural pathways. The etiology is likely multifactorial, with contributions from all three theories [1].
Associated Malformations
Laryngomalacia is seen in otherwise healthy children and in those with a wide range of comorbidities, including neurologic disease, cardiopulmonary disease, congenital anomalies, and syndromes. Up to 80 % of children with laryngomalacia have gastroesophageal reflux disease (GERD). Synchronous airway lesions have been reported in 12–64 % of children with laryngomalacia [8, 9]. Of these lesions, subglottic stenosis, tracheomalacia, and vocal cord paralysis are the most common [5, 8–10].
Clinical Features
Laryngomalacia is characterized by the onset of high-pitched inspiratory stridor within the first two weeks of life. Stridor is exacerbated by feeding, crying, and lying in a supine position. Symptoms typically peak around 6 months of age, and spontaneously resolve by 12–24 months. Symptoms range from mild inspiratory stridor and feeding difficulties to life-threatening airway obstruction, and cardiopulmonary complications. In severe cases, apneic spells, cyanosis, and failure to thrive may result [3]. Classification of laryngomalacia is described based on the site of supraglottic obstruction [8] or symptoms and clinical findings [1]. We currently classify laryngomalacia based on clinical findings.
Diagnosis
Transnasal flexible fiberoptic laryngoscopy is the gold standard for diagnosis of laryngomalacia. Characteristic findings include short aryepiglottic folds (100 %), floppy cuneiforms (60 %), and an omega-shaped epiglottis (20 %). Figure 6.4 demonstrates schematic illustrations and endoscopic photographs of airways of infants who have laryngomalacia. Figure 6.4a, c represents the more common finding of anterior prolapse of the epiglottis and aryepiglottic folds. Figures 6.4b, d represent the less common form, whereby the mucosa over the arytenoid and cuneiform cartilages and aryepiglottic folds are redundant. In the latter case, the children may present with a lower-pitched purring or rattling stridor on inspiration.
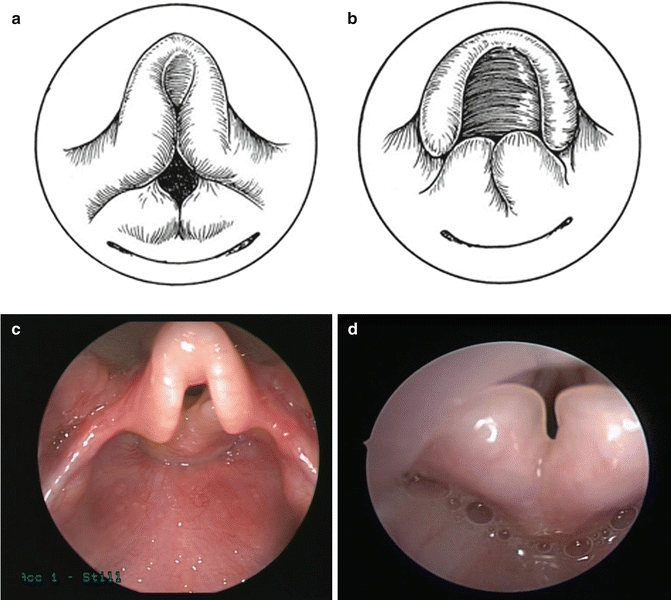
Fig. 6.4
(a and c) Illustration and endoscopic photograph demonstrating the most common form of laryngomalacia (anterior prolapse). (b and d) Illustration and endoscopic photograph demonstrating less common posterior prolapse
Adjunctive studies, including airway films and airway fluoroscopy, may be performed in patients whose symptoms are inconsistent with flexible laryngoscopy findings. Microlaryngoscopy and bronchoscopy are reserved for patients with suspected synchronous airway lesions or for those requiring surgical intervention for laryngomalacia.
Management
The management of laryngomalacia depends on individual clinical findings and disease progression. Most patients are monitored closely and do not require surgical intervention. Patients are managed medically with anti-reflux therapy. Parental reassurance should also be given. Surgical intervention is required in up to 20 % of patients with laryngomalacia [1]. Indications for operative management of severe cases include failure to thrive, hypoxia, pulmonary hypertension, and cor pulmonale. Supraglottoplasty is performed with laser or microlaryngeal instruments, with the aim of surgery being to release the tight aryepiglottic folds and reduce redundant supra-arytenoid tissue (Fig. 6.5). This approach can be tailored to individual laryngeal pathology. Tracheotomy is reserved for children in whom supraglottoplasty has failed, usually those with significant comorbidities. In children with severe neurologic deficits, tracheotomy may be an alternative to supraglottoplasty.
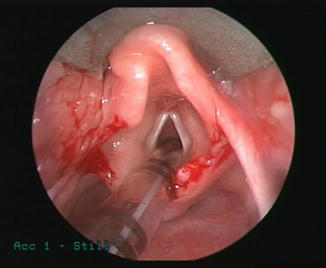
Fig. 6.5
Endoscopic photograph demonstrating postoperative results following supraglottoplasty performed to treat severe laryngomalacia
Laryngeal Cysts
Definition
Congenital laryngeal cysts are fluid- or air-filled lesions that cause variable degrees of hoarseness, dysphagia, stridor, and airway obstruction. Saccular cysts, laryngoceles, vallecular cysts, and lingual thyroglossal duct cysts are the lesions most commonly described.
History
Although the first description of laryngeal cysts is not well documented, the first classification of cystic laryngeal lesions was proposed by De Santo et al. in 1970 [11]. Arens et al. subsequently used the location of the cyst and histomorphology to describe laryngeal cysts [12]. More recently, Forte et al. have classified congenital laryngeal cysts by the extent of the lesion and the embryonic tissue of origin [13].
Incidence
The incidence of congenital laryngeal cysts has not been reported; however, these lesions are considered rare causes of airway and swallowing symptoms in the newborn.
Heredity
The mode of inheritance is unknown.
Etiology
The etiology of laryngeal cysts varies, depending on the type of cyst. Saccular cysts and laryngoceles arise from the saccule, a vestigial laryngeal structure. The saccule contains mucous glands thought to help lubricate the vocal cords. Congenital obstruction of the saccular orifice results in the development of a mucous-filled cyst. A laryngocele is an abnormal dilation of the saccule that maintains patency with the laryngeal lumen. This communication with the laryngeal lumen leads to an intermittent air-filled sac that causes variable airway symptoms. Vallecular cysts are thought to arise from obstruction of mucosal glands in the vallecula or base of tongue. The etiology of lingual thyroglossal duct cysts relates to the embryology of the thyroid. The thyroglossal duct is an epithelial-lined structure connecting the foramen cecum to the thyroid as it descends in the neck. Failure of this duct to obliterate between the fifth and tenth weeks of gestation can lead to the formation of a cyst anywhere along its tract; this can include the base of tongue, larynx, or neck.
Associated Malformations
There is no evidence that laryngeal cysts are associated with other anomalies or are more common in specific syndromes or conditions.
Clinical Features
Congenital laryngeal cysts present with airway symptoms and feeding difficulties. In neonates, any of these cysts may be life-threatening. Saccular cysts may present with life-threatening airway obstruction or gradual onset of stridor, depending on the size of the lesion at birth (Fig. 6.6). Laryngoceles typically cause intermittent symptoms including hoarseness and dyspnea made worse by crying (Fig. 6.7). Vallecular cysts often cause early onset of stridor associated with cough and cyanotic episodes (Fig. 6.8). Swallowing difficulties and failure to thrive may also occur. Symptoms related to thyroglossal duct cysts depend on cyst location and can include all of the above.
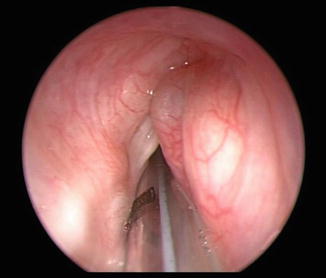
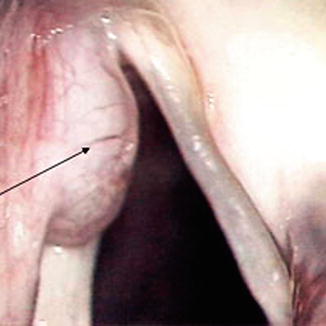
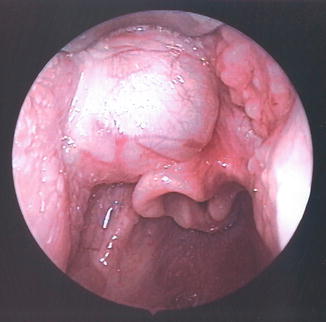
Fig. 6.6
Endoscopic photograph demonstrating an obstructing anterior-based saccular cyst
Fig. 6.7
Endoscopic photograph demonstrating a laryngocele of the larynx
Fig. 6.8
Endoscopic photograph demonstrating a vallecular cyst
Diagnosis
The gold standard for diagnosing laryngeal cysts is the use of transnasal flexible fiberoptic laryngoscopy. A soft-tissue lateral radiograph may be part of the initial evaluation prior to otolaryngology referral. Computed tomography (CT) can be useful in confirming the diagnosis and determining the extent of the lesion. Microlaryngoscopy and bronchoscopy are used at the time of surgery to evaluate the possible presence of secondary airway lesions and to secure the airway. Biopsy helps differentiate thyroglossal duct cysts from vallecular cysts by the presence of thyroid follicles.
Diagnostic investigations help to classify these lesions, allowing for appropriate treatment. Saccular cysts are classified as lateral (involving the false cord and aryepiglottic fold posterosuperiorly) or as anterior (protruding from the ventricle into the laryngeal lumen). Laryngoceles are considered internal if they are confined to the larynx and involve the false vocal cord and aryepiglottic fold (Fig. 6.9). External lesions extend beyond the thyrohyoid membrane.
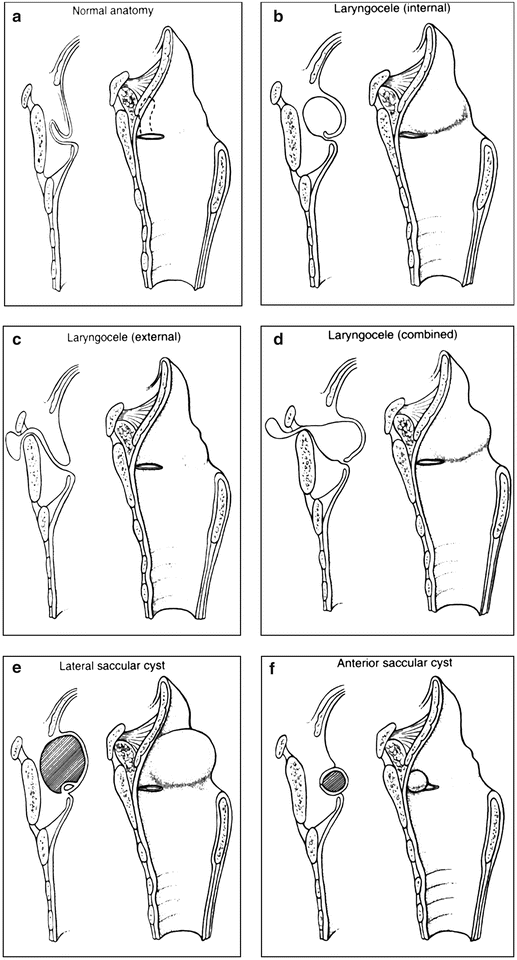
Fig. 6.9
Illustrations demonstrating the differences between saccular cysts and laryngoceles
Management
Treatment of laryngeal cysts is surgical. The diagnosis of saccular cysts is often confirmed with aspiration at the time of surgery. We advocate a lateral cervical approach as the primary surgical intervention. Temporary tracheotomy placement may be required for large life-threatening cysts. Laryngoceles may be treated endoscopically or through an external approach. Vallecular cysts are managed definitively with endoscopic marsupialization, or excision. We recommend that lingual thyroglossal duct cysts be endoscopically excised. Recurrences may require an external approach.
Laryngeal Webs and Atresia
Definition
Laryngeal webs are malformations in which abnormal tissue forms between two structures in the larynx [14]. True anterior glottic webs are a gossamer-thin membrane that connects the anterior true vocal cords; however, most laryngeal webs are actually thick, and involve not only the vocal cords but also the subglottis. These webs should be considered a milder version within the spectrum of laryngeal atresia.
History
Incidence
Although the incidence of laryngeal webs is unknown, anterior glottic webs account for 5 % of congenital laryngeal anomalies. More than 90 % of congenital laryngeal webs are anterior glottic webs; less than 2 % are found in the supraglottis; and 7 % are found in the subglottis. Complete laryngeal atresia is extremely rare.
Heredity
More than 50 % of children with partial laryngeal atresia have chromosome 22q11.2 deletion, an autosomal dominant heritable condition that usually presents as velocardiofacial syndrome (VCFS) or Di George syndrome. As the clinical manifestation of VCFS may be subtle, we recommend that all children with partial laryngeal atresia undergo genetic testing for chromosome 22q11.2 deletion [16]. It should, however, be noted that the incidence of partial laryngeal atresia in children with VCFS is low.
Etiology
Laryngeal webbing or atresia results from a failure in embryogenesis. The primitive laryngeal aditus becomes a t-shaped opening by the growth of three masses that later form the epiglottis and two arytenoid cartilages. As they develop during the fifth to seventh weeks of gestation, the lumen obliterates. Failure of the lumen to recanalize by week 10 of gestation leads to varying degrees of obstruction in the form of webs or atresia.
Associated Malformations
The most common conditions associated with laryngeal atresia are VCFS and DiGeorge syndrome [16]. Glottic and supraglottic atresia are also associated with other foregut and non-foregut anomalies. These include esophageal atresia, tracheoesophageal fistula (TEF), limb defects, and urinary tract abnormalities [17].
Clinical Features
Thin anterior glottic webs may be asymptomatic or present with mild voice dysfunction and hoarseness. Severe webs that are thick and involve the anterior and posterior glottis are more likely to present with biphasic stridor (particularly when infants are feeding or upset), life-threatening airway compromise, and aphonia. Infants are, nevertheless, remarkably tolerant of congenital airway compromise. They may present with subtle airway symptoms and yet have moderate to severe webbing [18]. Complete laryngeal atresia is not compatible with life unless ventilation through a TEF is achieved or the pathology is noted prior to birth and an EXIT (ex utero intrapartum treatment) procedure is performed to allow tracheotomy to be performed while maternal-fetal circulation is maintained.
Diagnosis
Initial evaluation is made by awake transnasal flexible fiberoptic laryngoscopy. This may suggest the need for rigid microlaryngoscopy or bronchoscopy in the operating room to confirm the diagnosis. The degree of webbing is noted and categorized according to the classification described by Cohen [15]. Type 1 webs are anterior and thin, occupying 35 % or less of the glottis. Type 2 webs involve 35–50 % of the glottis and have a thicker anterior component (Fig. 6.10). Type 3 webs are again anterior and occlude 50–75 % of the glottis. The thick anterior component may be associated with cricoid cartilage involvement and the vocal cords may not be seen. Type 4 webs are thick anteriorly and posteriorly, compromising 75–90 % of the glottis; there is a subglottic component and the vocal cords are not discernible (Fig. 6.11). Isolated posterior glottic webs are typically thin, but may have interarytenoid involvement with resultant vocal cord fixation. Vocal cord mobility should be documented during microlaryngoscopy and bronchoscopy. Imaging studies, including high-voltage lateral radiographs and CT are useful to help diagnose cricoid involvement or other airway anomalies.
Fig. 6.10
Endoscopic photograph of a Cohen type 2 anterior glottis web
Fig. 6.11
Endoscopic photograph of a Cohen type 4 glottic web
Laryngeal atresia can be diagnosed prenatally by ultrasound. Obstruction of the laryngeal lumen can result in congenital high airway obstruction syndrome (CHAOS). Signs of CHAOS are seen on ultrasound and include a flattened diaphragm, a fluid-filled, dilated airway distal to the obstruction, fetal hydrops, and enlarged hyperechogenic lungs. These signs indicate the need for color-flow Doppler to localize the level of obstruction [19, 20].
Management
Treatment for laryngeal webs involves either endoscopic or open surgical techniques that are typically delayed until 3–4 years of age. Thin anterior webs can be treated endoscopically with microlaryngeal instruments. Silastic sheeting or keels can be placed endoscopically or with open techniques to prevent stenosis. In this setting, a tracheotomy is usually placed to ensure a safe airway.
Webbing that involves the subglottis and cricoid plate requires a formal laryngotracheoplasty with laryngofissure. The CO2 laser is not recommended because it can cause scarring. The web is divided and the cricoid is addressed by submucosal resection of the cricoid cartilage, the use of anterior cartilage grafting, or partial cricotracheal resection. This can be performed with the use of a keel and a temporary tracheotomy or in a single-stage fashion with intubation for up to 2 weeks. Revision surgery is often required. Alternatively, open reconstruction of the anterior commissure can be performed when at least 25 % of the laryngeal lumen is patent. Immediate surgical intervention with a tracheotomy at birth may be necessary for type 3 and type 4 webs. Posterior glottic webs often can be addressed by endoscopic division, but may require open placement of a posterior costal cartilage graft for interarytenoid involvement.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


