Chapter 31 Genetic Mechanisms of Retinal Disease
Introduction
The purpose of this chapter is to provide an overview of concepts underlying our current understanding of the genetic basis of inherited retinal diseases (iRDs). iRDs are perhaps the best understood of human hereditary disorders. In part this is because diseases that affect vision are easily recognized and the retina is an accessible and well-characterized tissue. In many ways, though, we are still at an early stage of understanding the causes and consequences of these diseases. In fact, the causes of iRDs are highly varied: many different types of retinal disease are known, many different genes are involved, and there may be dozens of disease-causing mutations reported within a single gene. For example, currently at least 220 genes are known which can cause one or another form of retinal disease,1 and over 5000 mutations have been reported, in total, in these genes.2 In spite of the underlying complexity, it is now possible to identify the disease-causing gene and mutation, or mutations, in a substantial fraction of affected individuals and families.3,4
Therefore the cause of disease in an individual with an inherited condition such as RP is “simple,” in the sense that only one gene is affected (and usually affected in an obvious way), whereas there may be multiple contributory factors in an individual with AMD and the differences may be subtle. We already known exceptions to this rule – for example, there are digenic forms of RP with two affected genes5 – but the exceptions are rare.
This chapter focuses on genetic differences that are single-gene in nature and have a direct cause-and-effect relationship with disease, that is, inherited diseases of the retina. Genetic factors contributing to AMD are discussed in Chapter 64 (AMD: Etiology, genetics, and pathogenesis).
Basic concepts in human genetics
Inheritance
Figure 31.1 shows pedigrees illustrating autosomal dominant, autosomal recessive, and X-linked recessive inheritance (see Nussbaum et al.6 for details).
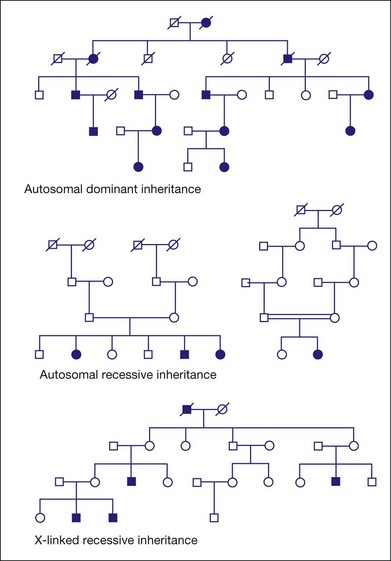
Fig. 31.1 Pedigrees illustrating autosomal dominant, autosomal recessive, and X-linked recessive inheritance.
Autosomal dominant inheritance
Variable clinical expression means that individuals with the same mutation may vary in onset, progression, or severity of disease or, in some cases, may have distinctly different clinical findings. Autosomal dominant RP is notoriously variable in expression. For example, mutations in one autosomal gene, PRPH2 (also known as RDS), can cause dominant RP, dominant macular degeneration, or dominant panretinal maculopathy, even among members of the same family.7–11
Incomplete penetrance, or nonpenetrance, means that some individuals with a disease-causing mutation will not be affected. For instance, 20% of individuals with a dominant-acting mutation in PRPF31 will have normal vision by age 60 even though relatives with the same mutation may have RP by age 20.12–15 One indicator of nonpenetrance in a multigenerational family is a “skipped generation,” that is, an unaffected individual with an affected parent and an affected child. This is often seen in families with PRPF31 mutations.
Autosomal recessive inheritance
Examples of autosomal recessive retinopathies include Leber congenital amaurosis and Usher syndrome.
X-linked or sex-linked inheritance
The disease status of female carriers is more complex, though. Although females have two Xs, one of the Xs, selected at random in each cell, is inactivated in most tissues. This is X-inactivation or lyonization, named for Mary Lyon, who first described the phenomenon.16,17 Lyonization increases the likelihood that a female carrier will be affected since some cells will express only the mutant protein. In fact, many female carriers of X-linked RP mutations show clinical symptoms. Females are less severely affected than males with the same mutation, but female carriers of X-linked RP mutations may have significant loss of vision by midlife or earlier.18–22
One consequence of clinical disease in carrier females is that families with X-linked RP may appear to have autosomal dominant RP if several females are affected.23 This is an example of complexities that arise in determining the mode of inheritance of iRDs.
Digenic and polygenic inheritance
Nearly all iRDs are monogenic, with only one gene affected per person. This is based on empirical observation, but it may be misleading since more complex forms of inheritance are hard to prove. Two counter examples are known for iRDs. First, one form of RP is caused by a combination of one mutation in the PRPH2 (RDS) gene and another mutation in the ROM1 gene.5,22 These two mutations are benign alone but pathogenic in combination. This is digenic inheritance. Secondly, Bardet–Biedl syndrome (BBS), a form of RP combined with congenital abnormalities, is in most instances a recessive disease with mutations in any one of at least 15 known BBS genes.1,24 Some cases of BBS, though, require a third mutation in a second BBS gene for disease expression.25,26 This is called trigenic or triallelic inheritance. Whether these examples of polygenic inheritance of iRDs are just rare anomalies or hint at greater complexity of retinal diseases is unclear.
DNA, RNA, and proteins
Figure 31.2 shows the steps in DNA duplication, RNA translation, and protein synthesis.27
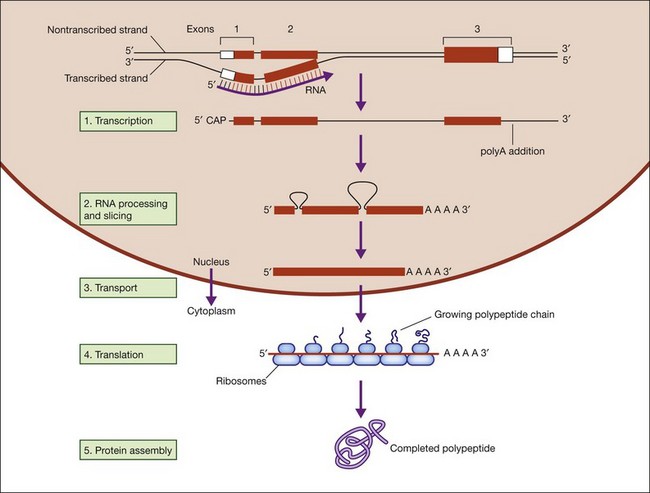
Fig. 31.2 Steps in DNA duplication, RNA translation, and protein synthesis.
(Reproduced from Nussbaum RL, McInness RR, Willard HF. Thompson and Thompson’s genetics in medicine, 7th ed. Philadelphia, PA: Saunders Elsevier; 2007, p. 31, with permission from Elsevier.)
DNA function is called the central dogma of DNA in recognition of the landmark explanation of DNA structure and function by Watson and Crick in 1953, and subsequent unraveling of the genetic code over the next decade.28,29 DNA is comprised of a phosphate backbone with nucleotide bases, A, T, G, or C, in linear array along the backbone. The backbone is conventionally drawn from the 5’ phosphate on one end to the 3’ phosphate on the other end. The opposite strand forms by pairing of cognate bases, A to T and G to C, on the parent strand. The opposite strand naturally aligns in a helical, antiparallel fashion, from 3’ to 5’ phosphates. This arrangement essentially explains inheritance in all living things.
Gene structure
Figure 31.3 shows gene structure based on the relationship between the protein sequence, mRNA intermediate, and original DNA gene sequence.27
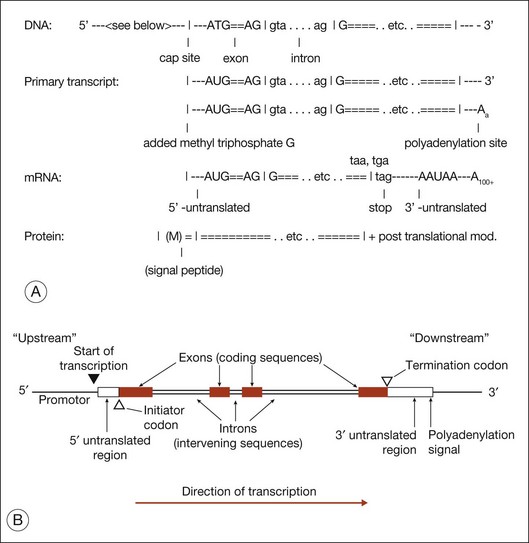
The evolutionary significance of splicing is still disputed, but its functional consequence is clear: it vastly increases the number of distinct proteins. This is because when splicing occurs, alternate combinations of introns may be removed. Alternate splicing is the norm in human genes, not the exception, and usually results in alternate mRNAs and alternate protein isoforms – all from a “single” gene. There are many examples of alternately spliced retinal genes producing multiple protein isoforms.30,31
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


