Fig. 2.1
Morphogenesis of the spiral ganglion. The spiral ganglion develops from a proneurosensory region (dark gray) in the anteroventral quadrant of the otic cup, beginning around E9 in mouse. Over the next week, the otic cup invaginates and acquires its mature three-dimensional structure. In parallel, the neurons delaminate to form a cochlear-vestibular ganglion (CVG). The vestibular (blue) and spiral (purple) ganglia gradually separate and eventually innervate the vestibular and auditory sensory epithelia respectively. The geniculate ganglion (tan) is initially attached to the CVG laterally (shown only for E10.5). This ganglion is separated from the CVG by the facial nerve and originates from a distinct placode
Inner ear neurons develop from precursors in the anteroventral quadrant of the otic vesicle, which leave the epithelium and proliferate to form the cochlear-vestibular ganglion (CVG) just rostral to the developing inner ear. At this stage, the cochlear and vestibular ganglia are distinct yet closely associated with each other, as well as with the geniculate ganglion (Fig. 2.1), forming a three-ganglion complex. Morphologically, neuroblasts can be recognized during otic cup stages as otic epithelial cells that lose their columnar morphology and delaminate from the epithelium (Carney & Silver, 1983; Hemond & Morest, 1991b), forming a distinct CVG by the 22–24 somite stage in mice (Wikstrom & Anniko, 1987), with the auditory division positioned medial and ventral to the vestibular division and the entire neural anlage still attached to the geniculate ganglion (Sher, 1972). The spiral ganglion grows and extends together with the cochlear duct over the next several days (Carney & Silver, 1983), gradually separating from the vestibular and geniculate ganglia.
The basic sequence of events during spiral ganglion development is conserved across species, with a few notable exceptions. In every species examined, neurons seem to be the first differentiated cell type to appear in the inner ear. In both mice and birds, neurogenesis begins during otic cup stages and continues after the otic vesicle has closed and become free from the overlying ectoderm (Carney & Silver, 1983; Hemond & Morest, 1991a). However, the structures for hearing exhibit distinct forms. In mammals, the auditory sensory epithelium (the organ of Corti) spirals within a coiled cochlear duct. Birds (and reptiles) instead detect sound via the basilar papilla, which is located in the tube-like lagenar recess. To underscore its similar function, the avian hearing organ is often referred to as the cochlear duct, but it is important to note that these structures are not strictly analogous, as the lagenar recess also houses an additional sensory epithelium, the lagena macula, that is not present in the cochlea.
The lagena macula is a sensory organ found in birds, fish, reptiles, and amphibians but not in mammals (Harada et al., 2001). Most often considered vestibular in nature, its function remains unclear and may vary among species (Fritzsch & Straka, 2014). In pigeons, there is intriguing evidence for a role in magnetoreception, which is the ability to orient in response to the Earth’s magnetic field (Wu & Dickman, 2011). From an evolutionary point of view, the lagena macula seems to have been lost from mammals, possibly having been incorporated into the apex of the organ of Corti, which itself evolved from the basilar papilla (reviewed in Fritzsch et al., 2013). Because of these differences, auditory and vestibular neurons form distinct ganglia in mammals but remain as a CVG in birds. Whether this difference in organization affects the development of the auditory neurons is not clear, but the presence of the lagena macula should be borne in mind when making comparisons between species, especially because this sensory epithelium is innervated by a subset of neurons that project to distinct targets centrally (Mahmoud et al., 2013).
The fish inner ear also exhibits a number of salient differences from both the avian and mammalian ears. Most strikingly, fish such as the common model organism zebrafish (Danio rerio) do not develop a cochlea-like structure and have no basilar papilla, as fish rely instead on hair cells in the saccule and lagena for auditory function (Bigelow, 1904). This raises the question of whether the neurons that innervate the saccule and lagena in fish are more analogous to vestibular or auditory neurons in mammals. Circuit tracing studies have confirmed that the primary sensory neurons that innervate each of these structures convey information to different regions of the brain, suggesting that fish use the information from sound-induced vibrations in the saccule and lagena differently from the movement-induced vibrations of the utricle and semicircular canals (McCormick & Wallace, 2012). However, in many brainstem nuclei, there is also considerable overlap with projections that are vestibular in nature. In addition to the murky understanding of auditory versus vestibular identity, the timing of neurogenesis is also slightly different in fish. Whereas in birds and mammals, hair cells typically develop after neurons, both populations are produced at the same time in fish (Haddon & Lewis, 1996) and continue to be added long after hatching (Popper & Hoxter, 1984). Fish also differ in the size of the ganglion, which contains only a few hundred neurons (Popper & Hoxter, 1984) compared to approximately 8000 in mice (Johnson et al., 2011) and approximately 9000 in chickens (Ard & Morest, 1984).
The human spiral ganglion is even larger, with approximately 30,000 neurons (Rasmussen, 1940; Nadol, 1988; Spoendlin & Schrott, 1989), although it appears to pass through developmental stages similar to what has been described in mice and chickens (Streeter, 1906; Bibas et al., 2006; Locher et al., 2013). In addition, there is accumulating evidence that the same basic pathways operate in fish, mice, and chickens, though different specific players may be involved in each species (see Groves & Fekete, 2012 for review). Much less is known about the molecular basis of human spiral ganglion neuron development, but to date, no obvious differences have been described. Nevertheless, it is important to keep in mind that differences between species may exist, both among model organisms and between model organisms and humans. Because of the closer parallels to the human system, findings from chicken and rodents will be emphasized here. The chick has served as an excellent system for working out the earliest stages of neurogenesis because of its accessibility for acute embryological manipulations. Mice offer the advantage of genetics plus closer parallels to the human.
Morphogenesis of the spiral ganglion depends on a coordinated series of extrinsic and intrinsic patterning events that begin in the early otic vesicle with the production of multipotent neurosensory progenitors. Neurosensory progenitors are progressively directed toward the spiral ganglion neuron (SGN) fate through a series of fate decisions. In parallel, the number and location of SGNs is controlled by selective expansion and culling of progenitor populations as well as directed migration away from the otic vesicle into the surrounding mesenchyme. As a result of these developmental events, the mature cochlea houses a population of SGNs that have both the intrinsic properties and the precise connections necessary for accurate transmission of sound information from hair cells to the central nervous system. Building on decades of careful anatomical and embryological studies, we now have a broad understanding of how these events unfold at the cellular level, and have begun to identify many of the signaling pathways and transcriptional networks that initiate and regulate early SGN development.
2.2 Origin of Inner Ear Neurons
2.2.1 The Otic Vesicle
In all species, neurogenesis is confined to spatially restricted regions of the otic cup, partially overlapping with zones that produce the sensory cells but excluded from those fated for non-neurosensory tissues in the mature inner ear. In rodents and birds, the neurons of the inner ear arise in the anteroventral quadrant of the otic vesicle (Figs. 2.1 and 2.2). Although intuited from histological studies, which revealed an obvious region of delamination (Knowlton, 1967; Carney & Silver, 1983), in vitro fate mapping studies ultimately confirmed that this portion of the otocyst produces neurons when dissected and cultured in isolation (Li et al., 1978; Adam et al., 1998), but that more dorsal and more posterior regions do not (Li et al., 1978). Similarly, dye labeling of the chick otic cup (Hamburger Hamilton [HH] stage 12) showed that CVG neurons are produced from the anterior compartment (Abello et al., 2007). Moreover, cells in this region rarely mixed with cells in the neighboring “non-neuronal” compartment and respected gene expression boundaries, indicating that the region of neurogenesis is patterned at an early stage.
The neurogenic zone itself appears to be further patterned, as vestibular neurons are generated before auditory neurons and from spatially distinct populations, as evidenced by dye labeling experiments in chicks (Bell et al., 2008) and genetic tracing experiments in mice (Koundakjian et al., 2007). Indeed, auditory and vestibular neurons appear to delaminate from different regions of the otic vesicle, with vestibular neurons developing close to the vestibular sensory epithelia and most auditory neurons instead delaminating from the boundary between the cochlea and saccule and then from the middle and apical turns of the cochlea itself (reviewed in Yang et al., 2011). Such spatial segregation is particularly extreme in fish, in which there are two separate neurogenic zones, with the anterior region producing neurons that innervate the utricle and the posterior region producing neurons that innervate the saccule, with anterior neurogenesis occurring slightly earlier (Haddon & Lewis, 1996; Haddon et al., 1998; Sapede & Pujades, 2010).
Importantly, in each species, the neurogenic zone is closely associated with regions of the epithelium that produce sensory cells, namely the hair cells and supporting cells of the utricle, saccule, cristae, and cochlea. Hence, the neurogenic zone is actually contained within a larger “proneurosensory domain” (PNSD) that contains both neural and sensory progenitors, with the neurogenic region overlapping with the nascent sensory epithelia for the utricle and saccule (Cole et al., 2000; Raft et al., 2007). Other sensory areas, such as those for the cristae, can arise either from apparently non-neurogenic regions in the PNSD or outside of the PNSD altogether, as suggested by expression of sensory markers and fate mapping (reviewed in Fekete & Wu, 2002).
2.2.2 Other Potential Sources for SGNs
Although the otic vesicle is the primary source for inner ear neurons, there is a long history of studies considering the possibility of a contribution from the neural crest and/or neuroepithelium. In fact, Bartelmez argued strongly that auditory neurons derive from the neural tube based on his histological analysis of early human embryos (Bartelmez, 1922). In contrast, early embryological experiments using the larval salamander indicated that most if not all neurons derive from the otic placode, whereas the neural crest produces the Schwann cells that myelinate the inner ear neurons (Yntema, 1937). Similarly, when neural crest cells were transplanted from quails to chicks, many quail-derived glia populated the mature ganglia and eighth nerve, confirming that auditory Schwann cells share the same neural crest origin as those in the rest of the peripheral nervous system (D’Amico-Martel & Noden, 1983). More surprisingly, some quail-derived cells also appeared to develop as neurons, but these seemed more likely to be vestibular based on their location. However, it was not possible to rule out a contaminating non-neural crest cell population, nor were markers used to verify the neuronal identity of the quail-derived cells.
More recently, an argument for a neural crest contribution was made based on genetic fate mapping studies in mice (Freyer et al., 2011). In these experiments, neural crest cells were permanently marked by Cre-mediated recombination of fluorescent reporters. In mice that produce Cre under the control of the Pax3 promoter, which is active throughout the dorsal neural tube, fluorescent cells could be seen moving from the neural tube into the otic vesicle, eventually contributing to the ganglion, maculae, and cochlea. However, these types of experiments can be difficult to interpret, as even undetectably low and likely physiologically irrelevant levels of Cre protein might be sufficient to induce recombination. In addition, Pax3 expression is not restricted to the neural crest, so many of these cells may be occasional neuroepithelial precursors that accidentally found themselves in the otic cup, which is pressed up against the hindbrain, but were nonetheless able to proliferate and differentiate within this new environment. In fact, dye labeling of cells in the early embryonic chick hindbrain revealed a similar contribution to various tissues of the inner ear, including the CVG (Ali et al., 2003). Hemond & Morest, (1991b) further noted a possible contribution from migratory cells formed at the boundary of the otic cup in chicks. These so-called “otic crest” cells seemed to stream toward multiple ganglia, but a specific contribution to the CVG was not defined.
In fact, many experiments argue that any non-otic contribution to the neurons of the inner ear is minimal or even nonexistent. For instance, complementary fate mapping studies using multiple independent Cre lines with expression in the otic vesicle, namely Foxg1–Cre (Hebert & McConnell, 2000), Pax2–Cre (Ohyama & Groves, 2004), and Pax8–Cre (Bouchard et al. 2004), suggest that the vast majority if not all inner ear neurons do in fact derive from the otic epithelium. In addition, fate mapping with Wnt1–Cre as well as a more restricted Pax3–Cre driver did not reveal any substantial contribution to the inner ear besides Schwann cells (Sandell et al., 2014). The conflicting results for Wnt1–Cre could reflect variation in the level of Cre activity in combination with different reporters and on different genetic backgrounds. Because the currently available “otic” Cre lines also mediate recombination in the neuroepithelium, albeit within highly restricted regions (Hebert & McConnell, 2000; Bouchard et al., 2004; Ohyama & Groves, 2004), generation of a truly otic-specific Cre line may be necessary for final resolution of this issue. Nonetheless, most studies indicate that the otic vesicle does indeed serve as the primary source for inner ear neurons.
2.3 Overview of SGN Development
SGNs pass through a number of developmental stages, from their origin in the proneurosensory domain to their final differentiation and maturation within the cochlea. Following on the careful descriptive studies performed by embryologists and anatomists at the beginning of the 20th century, our understanding of how SGN development progresses has been greatly aided by the more recent identification of genes that are expressed in a subset of cells and that have been shown to affect specific features of inner ear neuronal development. The first step is the production of proneurosensory progenitors, which are recognized by expression of Lunatic fringe (Lfng) (Morsli et al., 1998; Cole et al., 2000), Sox2 (Kiernan et al., 2005; Neves et al., 2007), and fibroblast growth factor 10 (FGF10; Pirvola et al., 2000; Alsina et al., 2004). Subsequently, a subset of proneurosensory cells upregulate expression of Delta-like 1 (Dll1) and Neurogenin1 (Neurog1) (Adam et al., 1998; Ma et al., 1998; Alsina et al., 2004; Brooker et al., 2006). Neurog1-positive precursors begin to express Neurod1 as they delaminate from the otic vesicle (Liu et al., 2000; Kim et al., 2001; Bell et al., 2008) and quickly down-regulate Neurog1 (Evsen et al., 2013).
Delaminated neuroblasts continue to divide within the nascent ganglion, exiting the cell cycle along the basal-apical axis, starting in the mid-base region around E9.5 in mouse, with only a few neurons still dividing in the apical turn at E13.5 and none at E14.5 (Ruben, 1967; Matei et al., 2005). This general progression fits with Neurog1-based birthdating studies (Koundakjian et al., 2007), though it is important to bear in mind that onset of Neurog1 precedes cell-cycle exit, so different aspects of timing are measured by these two methods. Early neuroblasts express Islet-1 (Adam et al., 1998; Li et al., 2004), which is sustained beyond the transient expression of Neurod1 (Radde-Gallwitz et al., 2004; Deng et al., 2014). The delaminated neurons begin expressing first Pou4f1 and then, after becoming postmitotic, Pou4f2 (Deng et al., 2014). Maturation is also marked by production of βIII-tubulin (i.e., TuJ1) (Radde-Gallwitz et al., 2004; Bell et al., 2008), which is maintained as the neurons differentiate and coalesce in distinct vestibular and spiral ganglia.
As they mature, neurons extend processes back toward the otic epithelium (Carney & Silver, 1983). In mice, neurons mature first in the base, with neurites present along the path by E11.5 (Carney & Silver, 1983) and peripheral processes extending into the cochlea by E12.5 (Farinas et al., 2001; Koundakjian et al., 2007; Appler et al., 2013). In parallel, SGNs extend central axons out of the ear and toward the auditory brainstem to form the auditory division of the eighth cranial nerve. The axons reach the hindbrain by E11.5 and quickly bifurcate, extending an ascending process rostrally toward the developing anterior ventral cochlear nucleus and a descending process caudally toward the posterior ventral cochlear nucleus and dorsal cochlear nucleus (Lu et al., 2011). By E15.5, SGN peripheral processes have penetrated the cochlear duct along its entire extent, and the central processes are topographically organized within each division of the developing cochlear nuclei (Koundakjian et al., 2007). The arrival of peripheral and central axons at their targets coincides with a peak in cell death first in the vestibular system and then in the cochlea (Nishizaki et al., 1998; Farinas et al., 2001). A similar sequence of events occurs in chicks (Ard & Morest, 1984; Whitehead & Morest, 1985; Molea & Rubel, 2003).
In mammals, there are two clear subtypes of SGNs that can be distinguished based on morphology by late embryogenesis (Bruce et al., 1997; Koundakjian et al., 2007). Type I SGNs extend radial projections directly toward the inner hair cells, whereas a minority population of Type II SGNs instead grow toward the outer hair cells and spiral toward the base. Initially, all SGNs produce Peripherin, but this expression is eventually restricted to Type II SGNs at postnatal stages (Hafidi, 1998). SGNs are further classified based on their spontaneous firing rates (Liberman, 1980) and variation in firing properties and gene expression (reviewed in Davis & Liu, 2011), but to date there are no molecular markers for early Type II SGNs or any other well-defined SGN subtype.
Efforts to understand the molecular pathways that govern early SGN development have uncovered important roles for several extrinsic pathways that pattern the otic vesicle; promote neurogenesis; and control the specification, proliferation, and survival of committed SGN precursors. Many familiar signaling pathways are involved, often exerting distinct effects at different stages. Of particular importance are Notch signaling, as well as pathways activated by fibroblast growth factors (FGFs), bone morphogenetic proteins (BMPs), Sonic hedgehog (Shh), insulin growth factor (IGF), and the neurotrophins. In Sect. 2.4, the cellular and molecular events that govern SGN development are described, including the specific impact of relevant signaling pathways at each stage. Excellent reviews outlining the details of these pathways and their broader contributions to inner ear development are available for additional information (Wright & Mansour, 2003a; Varela-Nieto et al., 2004; Yang et al., 2011; Groves & Fekete, 2012; Kiernan, 2013; Neves et al., 2013).
2.4 Patterning the Proneurosensory Domain
Inner ear neurogenesis begins with the formation of the PNSD in the anterior otic cup during the earliest stages of development. As such, development of the PNSD is essentially a matter of establishing the anterior–posterior axis of the otic cup. The appearance of L–fng and other PNSD markers in the anterior otocyst is accompanied by a gradual restriction in the expression of nonsensory markers, such as Lmx1a/1b (Abello et al., 2007; Nichols et al., 2008; Vazquez-Echeverria et al., 2008) and Tbx1 (Raft et al., 2004), to the posterolateral domain as early as the 10–12 somite stage in mice (Fig. 2.2). Genetic fate mapping studies have confirmed that Tbx1-positive cells are excluded from the neurogenic zone (Xu et al., 2007). However, similar genetic fate mapping data for L–fng and FGF10 are not available, leaving open the question of the degree of fate restriction within the PNSD at this early stage.
2.4.1 Patterning Signals from Outside of the Inner Ear
Initial efforts to understand how the PNSD is patterned showed that the anterior–posterior axis is fixed remarkably early. For instance, anterior pieces of the chick otic epithelium are able to generate neurons in vitro, even in the absence of any surrounding tissues (Adam et al., 1998). Similarly, when otic cups were transplanted with a reversed orientation in HH stage 16 chick embryos, the PNSD remained in its original position (Wu et al., 1998). However, a similar manipulation between HH stages 10–12 caused the L–fng domain to form in what was originally the posterior half of the otic cup (Bok et al., 2005). Thus, signals from the surrounding tissue initially influence where the PNSD will form, but the axis is fixed just after the 16 somite stage (HH stage 12). In contrast, the dorsal–ventral axis remains sensitive to changes in orientation for longer, with important cues provided by the notochord, floorplate, and dorsal neural tube (Wu et al., 1998; Bok et al., 2005). Changes to the dorsal–ventral axis can also affect the position of the neurogenic domain (Bok et al., 2005; Riccomagno et al., 2005), highlighting the complexity of the interactions that ultimately shape the PNSD. However, the timing of events suggests that signals along the anterior–posterior axis provide the primary cues for PNSD formation.
Embryological experiments have helped narrow down the possible source of the cues that pattern the otic cup. The hindbrain does not appear to provide essential anterior-posterior information, as rotations prior to the 16 somite stage did not affect the location of the PNSD (Bok et al., 2005). Instead, signals seem to come from the nearby periotic ectoderm and somatic mesoderm (Bok et al., 2011). A major component of this signal is retinoic acid (RA), which prevents neurogenesis when ectopically expressed in chicks or mice. Conversely, blockade of RA signaling causes expanded neurogenesis. Further, RA and other components of the pathway are present and actively maintained in tissues surrounding the early otic cup in chicks. A similar role for RA has been described in zebrafish (Radosevic et al., 2011), suggesting this is an evolutionarily conserved mechanism, though differences among species likely exist in terms of the source of RA, how the gradient is established, and how the signal is interpreted. Indeed, the complex expression patterns and wide range of teratogenic effects across species indicate that RA can affect multiple aspects of inner ear development (reviewed in Romand et al., 2006).
Additional critical patterning information appears to be provided by FGFs. In chicks, FGF8 is expressed close to the otic territory prior to the onset of neurogenesis and can promote neurogenesis when expressed ectopically (Abello et al., 2010). Conversely, broad pharmacological inhibition of FGF signaling caused a loss of PNSD markers, accompanied by an expansion of the nonsensory marker Lmx1b. Similar alterations in BMP signaling had no effect on neurogenesis, but did influence Lmx1b expression, confirming a role in anterior–posterior patterning. The expansion of a nonsensory marker in the absence of an effect on neurogenesis indicates that FGF and BMPs may act independently to pattern the axis, with additional signals such as RA influencing the final outcome. For instance, as in chicks, an FGF ligand is required for anterior-posterior patterning in fish (Hammond & Whitfield, 2011), but some of these effects may be due to changes in expression of genes required for RA metabolism (Radosevic et al., 2011).
Whether FGFs regulate PNSD formation in mice remains unclear, due in part to differences in how and when neuronal development has been assessed. The PNSD appears to be present in FGF3 mutants, though the CVG is noticeably smaller once it forms (Hatch et al., 2007; Vazquez-Echeverria et al., 2008). However, loss of both FGF3 and FGF10 can cause expanded neurogenesis (Vazquez-Echeverria et al., 2008). Similarly, Neurod1-positive cells form in ectopic locations in Kreisler mutant mice, which suffer from hindbrain patterning defects including reduced expression of FGF3 and FGF10, and this phenotype can be partially rescued by restoration of either FGF. On the other hand, loss of the FGF2R (IIIb) receptor, which binds both FGF3 and FGF10, does not impair early CVG development (Pirvola et al., 2000; Pauley et al., 2003). However, because early PNSD development was not assessed, it is possible that a similar phenotype was missed in these animals (Pirvola et al., 2000). Altogether, the exact contribution of FGF3/FGF10 signaling is difficult to pinpoint due to the variability of reported double mutant phenotypes (Wright & Mansour, 2003b; Vazquez-Echeverria et al., 2008) and the fact that the phenotypes do not recapitulate what is seen in FGF2R mutants (Pirvola et al., 2000; Pauley et al., 2003). Why FGFs may antagonize neurogenesis in some contexts yet induce neurogenesis in others, as well as the specific contributions of various family members, will be important to work out in the future.
Although anterior–posterior signaling pathways function first, the final shape and size of the PNSD is also influenced by cues that pattern the dorsal–ventral axis. Shh from the notochord and floor plate provides a potent ventralizing signal that is necessary for normal expression of sensory and nonsensory markers (Brown & Epstein, 2011). However, loss of the PNSD from Shh mutant mice does not appear to be a direct effect on otic vesicle patterning, as otic-specific ablation of the Shh receptor Smoothened did not reproduce this effect. Instead, the change in PNSD gene expression appears to be secondary to expanded Wnt signaling from the neural tube, which is dorsalized in Shh mutants (Chiang et al., 1996; Riccomagno et al., 2005). In support of this idea, activation of Wnt signaling also inhibits neurogenesis (Ohyama et al., 2006; Brown & Epstein, 2011). However, importantly, Wnt signaling is not necessary for neurogenesis per se (Ohyama et al., 2006). Hedgehog proteins may play a more direct role during anterior–posterior patterning of the zebrafish otic vesicle, where they appear to work in parallel with—yet independently of—FGFs (Hammond et al., 2003, 2010; Hammond & Whitfield, 2011). The basis of these apparent differences among species is unclear but could reflect unique features of different types of vertebrate ears.
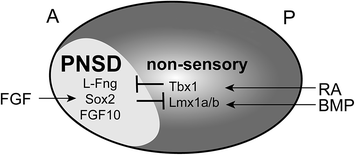
Fig. 2.2
Patterning the proneurosensory domain (PNSD). Extrinsic signals pattern the anterior–posterior (A-P) axis of the otic cup, with FGF inducing neurosensory development in the anterior and retinoic acid (RA) and BMPs inducing development of nonsensory structures in the posterior. As a result, PNSD markers such as L-fng, Sox2, and FGF10 are restricted to the anteroventral quadrant. These fate decisions are reinforced intrinsically by mutually antagonistic transcriptional networks, with Sox2 promoting the neurosensory fate and Tbx1 promoting the nonsensory fate
2.4.2 Intrinsic Patterning Mechanisms
Although signals extrinsic to the ear likely initiate the events that place the PNSD in the anterior otic cup, transcriptional networks and local cell–cell interactions play an important role in reinforcing these decisions and ensuring the neurosensory competence of cells within this domain. The transcriptional networks serve two complementary functions: to ensure expression of genes necessary for that cell’s needs at that point in time and to control the activity of the network itself. The Cell–cell interactions coordinate these intrinsic events with the surrounding tissue.
One important player in the PNSD is the transcription factor Tbx1, whose expression is restricted to the posterolateral (i.e. nonsensory) region of the otocyst as early as the 10 somite stage in mice (Raft et al., 2004). Neurog1 and Neurod1, which mark neural progenitors in the PNSD, are expressed in a complementary pattern to Tbx1 in the otocyst (Fig. 2.2). In mice with excess Tbx1, neurogenesis is reduced, particularly in the posterior of the PNSD (Raft et al., 2004; Freyer et al., 2013). Conversely, in Tbx1 mutant mice, Neurog1 and Neurod1 expression is increased (Raft et al., 2004; Xu et al., 2007), likely due to an expansion of the PNSD (Raft et al., 2004). Moreover, in Tbx1 mutants, cells from the Tbx1 domain populate the ganglion, something that never occurs in wild-type animals (Xu et al., 2007). Thus, Tbx1 actively represses the PNSD fate in nonsensory cells, thereby confining neurogenesis to a restricted domain. Extra Neurog1– and Neurod1-positive cells also develop in Tbx1 mutant fish (Radosevic et al., 2011), and spiral ganglion defects have been reported in human patients with velocardiofacial/DiGeorge syndrome (Schmidt, 1985), which is linked to TBX1 mutations (Yagi et al., 2003; Zweier et al., 2007). Hence, Tbx1 appears to play a basic, evolutionarily conserved role in determining where neurogenesis will occur.
If Tbx1 acts to prevent the neurosensory fate within the nonsensory region of the otocyst, then what factors promote this fate in the PNSD? The positively acting pathways are more complex, involving multiple transcriptional networks that interact with each other to drive production not only of neurons, but also of hair cells and supporting cells that will form the organ of Corti. One factor that appears to participate in many of these fate decisions is the SoxB1 family member Sox2. In mice, Sox2 is expressed in the ventral rim of the otic cup by E8.5 (Wood & Episkopou, 1999; Zou et al., 2008) and then in the ventrolateral otocyst at E9.5 (Mak et al., 2009). Subsequently, Sox2 can be detected both in delaminating neuroblasts and in the developing sensory epithelia (Kiernan et al., 2005; Mak et al., 2009). Sox genes show a similar expression in the PNSD in chicks, with expression complementing Tbx1 and Lmx1b by the 10 somite stage (Abello et al., 2007, 2010).
Just as Tbx1 promotes the nonsensory fate, so does Sox2 drive cells down the neurosensory path. In mice harboring mutations in Sox2, neurosensory development is severely disrupted, with an early loss of both the prosensory domain (Kiernan et al., 2005) and of neurons (Puligilla et al., 2010). The loss of both cell types suggests that Sox2 may be required for the initial specification of precursors in the PNSD. Moreover, in chicks, ectopic expression of Sox3 inhibits Lmx1b, hinting that mutually antagonistic interactions may solidify the nonsensory versus PNSD fate decision (Abello et al., 2010): wherever Tbx1 is on, Sox2 will be off and vice versa. Interestingly, Tbx1 expression was not affected. Similarly, Lmx1a but not Tbx1 shows reduced expression in Kreisler mutant mice, indicating there may be multiple parallel pathways that influence the nonsensory fate (Vazquez-Echeverria et al., 2008). Indeed, in Lmx1a mutant mice, Sox2 expression expands, followed by the formation of fused sensory epithelia and a larger CVG (Nichols et al., 2008; Koo et al., 2009).
As early acting factors such as Sox2 direct cells toward the neurosensory fate, additional transcriptional networks cooperate to determine whether individual progenitors will produce neurons, sensory cells, or both (Fig. 2.3). Unraveling the logic of this progressive fate restriction has been complicated by the fact that there seem to be multiple types of neurosensory progenitors within the PNSD. In favor of this idea, fate mapping in zebrafish has revealed three different types of precursors: neurosensory, neural, and sensory (Sapede et al., 2012). Similarly, in chicks, there is solid evidence that a common neurosensory progenitor produces both hair cells and neurons in the utricular maculae, but not in the other sensory organs, consistent with the presence of a heterogeneous precursor population (Satoh & Fekete, 2005).
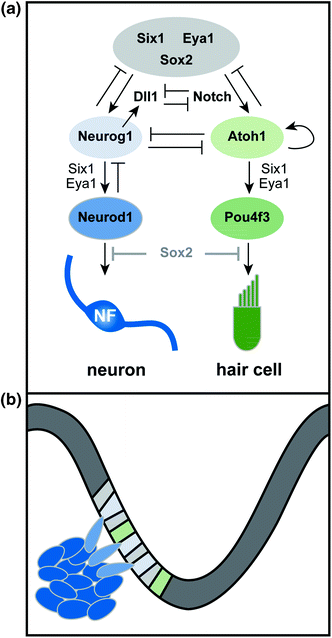
Fig. 2.3
Transcriptional control of neurogenesis. a Six1, Eya1, and Sox2 cooperate in PNSD progenitors (gray) to promote the neurosensory fate. Lateral inhibition mediated by Delta-like 1 (Dll1) and Notch steers neurosensory precursors toward either the neuronal (blue) or sensory (green) fate. In neural precursors, Neurog1 works with Six1 and Eya1 to promote differentiation, resulting in expression of Neurod1 and production of mature, neurofilament (NF)-positive neurons. Similarly, in prosensory progenitors (green), Atoh1 promotes its own expression and cooperates with Six1 and Eya1 to induce hair-cell differentiation pathways (i.e. Pou4f3) and production of hair cells. In some contexts, Neurog1 and Atoh1 show mutually antagonistic interactions. Downregulation of Sox2 occurs in differentiating neurons and hair cells; maintained expression of Sox2 interferes with maturation, suggesting that a decrease in Sox2 levels may be necessary for normal differentiation. b Diagram of neurogenesis in the context of the developing otocyst. Neuronal precursors are specified in the otic epithelium and then delaminate into the mesenchyme, where they continue to proliferate and then differentiate. Sensory precursors remain in the otic epithelium, where they ultimately produce both the hair cells and supporting cells of the organ of Corti (not shown)
The basis of the observed precursor heterogeneity remains unclear. One possibility is that hair cells and neurons derive from a similar early neurosensory progenitor that gradually shifts its potential over time, with the latest remnants of this population showing an extended ability to produce both hair cells and neurons in the maculae. Such a progenitor would be difficult to detect using standard fate mapping techniques. Alternatively, there may be a spatial segregation within the PNSD, with common progenitors limited to one subregion. Indeed, across species, proven bipotent progenitors exist in regions where the sensory domain, presaged, for example, by expression of BMP4, is found within the PNSD itself (Cole et al., 2000; Raft et al., 2007; Sapede et al., 2012).
Within the otocyst, local cell–cell interactions reinforce the intrinsic pathways that ultimately determine which cells will contribute to the neuronal lineage, influencing both the initial formation of the PNSD and the subsequent segregation of neuronal and sensory precursors. Both of these binary fate decisions appear to be under the control of the Notch pathway. As initially established in Drosophila, the Notch receptor interacts with a transmembrane ligand called Delta to mediate lateral inhibition and thereby promote the acquisition of distinct cell fates within a field of multipotent progenitors (reviewed in Schwanbeck et al., 2011). A second related ligand, Jagged, plays a similar role. In mammals, the basic pathway is conserved, with four Notch receptors, three Delta-like ligands, and two Jagged ligands. Ligand binding induces cleavage of the intracellular domain (ICD) of the Notch receptor protein in the neighboring cell. The Notch-ICD then enters the nucleus to directly regulate expression of target genes. Among the target genes are additional transcription factors that feedback to increase expression of Notch itself while simultaneously decreasing Delta production in that cell. The overall consequence is that neighboring cells ultimately express either Delta or Notch and therefore adopt one of two possible fates, for instance, whether to become a neuron. Because of the lateral inhibition mechanism, early uniform expression of the ligand and receptor is often followed by a more salt-and-pepper–like appearance within that field of cells, reflecting the gradual emergence of two distinct cell fates. Thus, activation of the Notch receptor by a Delta-family ligand offers a direct way to convert an extrinsic signal into an intrinsic change in gene expression.
Understanding the precise effects of Notch signaling in otic neurogenesis has been challenging because of the presence of multiple ligands and receptors, as well as differences in the effects of these molecules throughout development (reviewed in Kiernan, 2013). However, several lines of evidence suggest that Notch signaling promotes neurogenesis during inner ear development. For instance, like Lfng (Morsli et al., 1998), which is a known regulator of the Notch pathway, the Delta homolog Delta–like 1 (Dll1) is expressed in the PNSD at early stages (Abello et al., 2007; Daudet et al., 2007). In addition, pharmacological inhibition of Notch signaling in chicks can increase the number of Dll1+ cells (Daudet et al., 2007). Conversely, when Dll1 is absent in mice, too many neurons develop, consistent with a loss of lateral inhibition between Dll1+ neurons and the surrounding cells (Brooker et al., 2006). Interestingly, the production of extra neurons in Dll1 mutant mice apparently comes at the expense of the saccular and utricular maculae, whereas there is an increase in hair cell number in the cochlea, providing further evidence that there is a defined common neurosensory progenitor for only a subset of hair cells and neurons. Jagged ligands, on the other hand, may not be involved in inner ear neurogenesis (Zhang et al., 2000; Brooker et al., 2006; Neves et al., 2011).
One of the key consequences of Notch signaling is to induce expression of potent basic helix loop helix (bHLH) transcription factors, which act both to regulate Notch–Delta production and to induce cell-type–specific programs of gene expression. Similarly, whereas many genes are uniformly expressed in the PNSD, Dll1 shows a more irregular pattern, apparently reflecting upregulation in early neuronal precursors (Adam et al., 1998; Abello et al., 2007; Daudet et al., 2007) which express the proneural bHLH transcription factors Neurog1 and Neurod1. In Neurog1 mutant mice, no inner ear neurons form and Dll1 expression is lost, consistent with the classic model of lateral inhibition, with Neurog1 acting both to enhance production of Dll1 and promote the neuronal fate (Ma et al., 1998; Raft et al., 2007). The Dll1- cells, on the other hand, likely adopt a prosensory fate, which is promoted by a different bHLH transcription factor, Atoh1 (formerly Math1), which is required for hair cell development in mice (Bermingham et al., 1999). When signaling downstream of Notch is prevented, Neurog1 expression increases, as would be expected if Dll1 is no longer able to inhibit expression of Neurog1 in neighboring cells (Raft et al., 2007). In addition, pharmacological inhibition of Notch in chicks enhances the local upregulation of Dll1, indicating that lateral inhibition normally segregates Notch+ and Dll1+ populations within the PNSD (Daudet et al., 2007). Indeed, ectopic expression of the Notch-ICD is sufficient to create ectopic sensory regions and can divert neuroblasts to the hair cell fate in vivo (Daudet & Lewis, 2005; Hartman et al. 2010; Pan et al., 2010; Liu et al., 2012).
Because Neurog1 and Atoh1 are essential for neuronal and sensory development respectively, one attractive idea is that these two transcription factors participate in mutually antagonistic interactions within PNSD precursors that ultimately produce dedicated neuronal (i.e., Neurog1+) or sensory (i.e., Atoh1+) progenitors. If this model is true, then Neurog1 and Atoh1 must be coexpressed, at least at low levels, in any common neurosensory progenitor. This clearly appears to be the case in the utricle and saccule, where stripes of Atoh1 expression appear within the neurogenic domain (Raft et al., 2007). Moreover, descendants of Neurog1+ cells, as marked by genetic fate mapping, do indeed populate the maculae, exactly as predicted both by gene expression studies and viral fate mapping (Morsli et al., 1998; Cole et al., 2000; Satoh & Fekete, 2005). Additional evidence has come from analysis of Neurog1 and Atoh1 mutant mice: loss of Neurog1 is accompanied by an increased number of Atoh1-positive hair cell precursors in the developing utricular macula, whereas there are more neural precursors in the maculae of Atoh1 mutant mice (Raft et al., 2007). Ultimately, however, fewer hair cells develop in Neurog1 mutants, particularly in the saccule, indicating that the extra Atoh1-positive progenitors may not differentiate properly (Ma et al., 2000; Raft et al., 2007).
Together, these findings provide strong support for a common neurosensory progenitor in the maculae. Notably, the three major drivers of neuronal and hair cell fates in vertebrates—Neurog1, Neurod1, and Atoh1—are all bHLH factors that are closely related to each other and to bHLH factors with similar functions in sensory development in invertebrates. Indeed, it has been proposed that transient coexpression and cross-regulation of Neurog1, Neurod1, and Atoh1 may reflect an evolutionarily ancient series of interactions, with an expansion within the family eventually leading to a situation where individual bHLH factors segregate to neural versus sensory precursors (Pan et al., 2012).
The situation in the cochlea is less clear. First, Atoh1 expression does not appear to overlap with the neurogenic region of the cochlea (Raft et al., 2007). Accordingly, genetic fate mapping revealed no contribution of Atoh1-positive progenitors to the CVG (Yang et al., 2010). Conversely, the Neurog1 population does not produce any cochlear hair cells (Raft et al., 2007), despite massive labeling of the spiral ganglion neurons (Koundakjian et al., 2007; Raft et al., 2007). Nevertheless, there are some tantalizing phenotypes in the cochlea that suggest that Atoh1–Neurog1 interactions may occur, though perhaps only transiently. During normal development, neurons exit the cell cycle before hair cells (Matei et al., 2005). In contrast, in Neurog1 mutants, hair cell precursors exit the cell cycle prematurely (Matei et al., 2005), and extra rows of hair cells develop in some regions (Ma et al., 2000). This phenotype can be explained in part by the absence of SGNs, which normally produce a Shh cue that prevents hair cells from exiting the cell cycle (Bok et al., 2013). However, intrinsic effects might also contribute: when Neurog1 is lost, a progenitor that would normally become a neuron may instead select the alternate fate and differentiate as a hair cell (Matei et al., 2005).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


