Fig. 8.1
Schematic representation of the relationships between afferent and efferent auditory nerve fibers with two types of sensory hair cells. Spiral ganglion neurons (SGNs) are clustered in Rosenthal’s canal (RC) and include two groups of neuronal cells: large type I cells that make up 90–95 % of the SGN population and synapse with inner hair cells (IHCs) and small type II cells comprising 5–10 % of the neurons innervating outer hair cells (OHCs). The cell bodies of type II neurons are often seen in the periphery of RC, toward the osseous spiral lamina. Type I nerve fibers are surrounded by myelinating Schwann cells, whereas type II fibers are enclosed by nonmyelinating Schwann cells. Type I fibers lose their myelin sheath before they enter the organ of Corti through the habenular opening and can be classified into two (or three) populations based on their spontaneous discharge rate (SR). High-SR fibers are thick fibers with large terminals that contact the pillar side of IHCs. In contrast, low-SR fibers are thinner fibers with smaller terminals on the modiolar side of IHCs. Synapses on the modiolar side of IHCs have longer ribbons whereas synapses located on the pillar side of the cell have shorter ribbons. Radial innervations of efferent nerve fibers within the organ of Corti consist of (1) inner spiral fibers that run across the afferent nerve fibers under IHCs and (2) tunnel radial fibers that contact directly to OHC bodies with large nerve endings
There are two subpopulations of afferent neurons (types I and II) in the spiral ganglia of mammalian cochleas, each with their own morphological, immunostaining, and electrophysiological characteristics (Fig. 8.1; Davis and Crozier, Chap. 4). In most mammalian species (with the exception of humans), the cell bodies of type I SGNs are heavily myelinated. The remaining type II neuronal cells are unmyelinated and innervate the outer hair cells (OHCs) with a 1:10–20 ratio (Kiang et al., 1982; Liberman & Simmons, 1985). Both type I and type II neurons can be found within Rosenthal’s canal (RC). Their central projections form the auditory nerve within the internal auditory canal. Closely associated with the peripheral and central processes of these neurons are various glial cells, including Schwann cells, satellite cells, and oligodendrocytes. The peripheral portion of the auditory nerve is surrounded by myelinating Schwann cells (for type I SGNs) and nonmyelinating Schwann cells (for type II SGNs). The central portion of the auditory nerve is also enveloped by Schwann cells in the proximal part (before the glial transition zone) and by oligodendrocytes in the distal part (after the glial transition zone). The central projections of types I and II neurons form the modiolar segment of the auditory nerve, pass through the internal auditory canal and then enter the cochlear nucleus (Nayagam et al., 2011; Muniak et al., Chap. 6).
It is well established that auditory information from the cochlea is redundantly transmitted to the brain through type I SGNs that innervate each IHC (with about 20 synapses per IHC). The functional properties of type II neurons in transmitting auditory information are still largely unknown. Voltage- and current-clamp recordings of SGNs from postnatal rodents revealed rapidly inactivating A-type–like potassium currents in type II neurons (Jagger & Housley, 2003) and slow accommodation of responses to depolarization (Reid et al., 2004). In addition, recordings at the type II synapses show that the release of synaptic vesicles by OHCs results in a small-scale depolarization (Weisz et al., 2009). These results suggest that type II neurons are less active than type I neurons during normal auditory encoding processes. The selective survival of type II neurons has been seen in several cochlear and auditory nerve injury models. For example, pathological alterations of type I SGNs but not type II SGNs were seen after ototoxic lesions of sensory hair cells (Bichler et al., 1983; Leake & Hradek, 1988), noise trauma (Spoendlin, 1975; Lim, 1976), ouabain exposures (Lang et al., 2005), and transection of the cochlear nerve (Spoendlin & Suter, 1976). Future endeavors should address whether unique functional features of type II neurons make them less susceptible to injury in pathological conditions.
Type I afferent fibers are classified into two or three subgroups based on their spontaneous discharge rate and sensitivity to sound stimulation (Fig. 8.1 and Table 8.1). Auditory nerve fibers discharge spontaneously without stimulation. Spontaneous rate (SR) in auditory afferent fibers was first examined by Kiang et al. (1965) and then further defined into subgroups by Liberman (1978). In the cat, auditory nerve fibers are classified into three groups: low-SR (<0.5 spikes/s), medium-SR (0.5–18 spikes/s), and high-SR (>18 spikes/s) fibers. Similar SR-based functional subdivisions of auditory nerve fibers have also been reported in other mammalian species including chinchilla (Salvi et al., 1982; Frisina et al., 1996), guinea pig (Winter et al., 1990), gerbil (Schmiedt, 1989; Ohlemiller et al., 1991), and mouse (Taberner & Liberman, 2005). Auditory afferent fibers with higher SRs have low thresholds to stimuli, whereas fibers with lower SRs have higher thresholds (see Table 8.1). In addition, morphological evidence has shown that the specialization of central projections correspond to peripheral fibers based on their SR (review by Nayagam et al., 2011; Muniak et al., Chap. 6).
Table 8.1
Differential physiological and morphological characteristics of high-SR fibers and low- (and medium-SR) fibers
Differentiated characteristics | High-SR fibers | Low- (and medium-) SR fibers |
|---|---|---|
Physiological | ||
Spontaneous discharge rates | ||
Cat | 18–100 spikes/s | Low-SR: <0.5 spike/s Medium-SR: 0.5–18 spikes/s |
Gerbil | 18–150 spikes/s | <18 spikes/s for gerbil |
Mouse | 1–120 spikes/s | <1 spike/s |
Response threshold | Low | High |
Dynamic range | Smaller | Larger |
Threshold recovery following a prior stimulation | Faster | Slower |
Sensitivity to endocochlear potential | ~1 dB/mV | >1 dB/mV |
Morphological | ||
Peripheral terminal localization | Pillar pole of IHCs | Modiolar pole of IHCs |
Peripheral terminal | Larger | Smaller |
Ribbon | Shorter and thicker | Longer and thinner |
Receptor patch | Larger | Smaller |
Synapse vesicle | Less | More |
Axon diameter | Larger | Smaller |
Mitochondria within terminal | More | Less |
8.2 Loss of Spiral Ganglion Neurons and Their Processes
Many extrinsic and intrinsic factors can cause the degeneration and dysfunction of SGNs and their processes. These factors include exposure to noise and ototoxic drugs, infection, genetic defects, aging, and absence of auditory signaling input such as loss of sensory hair cells (Liberman & Kiang, 1978; Spoendlin, 1984; Zimmerman et al., 1995). Various loci of pathology in the auditory nerve with a list of representative references are included in Table 8.2. Loss of SGNs and their processes results in auditory impairment by reduction of the auditory information (e.g., timing, neural synchrony, and phase locking) delivered to the brain and by secondary degeneration in cochlear nuclei and other components of the central auditory system.
Table 8.2
A summary on various sites of degeneration reported in SGNs and their associated elements
Anatomic site with pathological changes | General characteristics of nerve dysfunction | Representative references |
|---|---|---|
1. Synapse | ||
Swelling and disruption of postsynaptic structure, reduced synapse ribbons, alterations of synapse location, orphan ribbon | Reduced activity, inexcitability, hyperexcitability (excitotoxicity), reduced activity of low-SR synapses, dys-synchronous auditory procressing, deficits in temporal coding, reduced suprathreshold amplitudes of auditory evoked potential | |
2. Peripheral process | ||
Loss or dysfunction of afferent fibers | Inexcitability, abnormal nerve activity, reduced activity of low-SR fibers, decreased suprathreshold amplitude of auditory evoked potentials | |
3. Neuronal cell body | ||
Reduced nuclear area neuronal apoptosis | Inexcitability, no conduction, reduced suprathreshold amplitude of auditory evoked potentials | |
4. Central axon | ||
Disintegration of myelin sheath, retrograde degeneration of axon | Decreased suprathreshold amplitude, inexcitability, no conduction | |
5. Myelin sheath | ||
Demyelination (axon survives for short period) | Slow nerve excitability, dys-synchronous, slow conduction velocity, long latency response | |
It is important to note that ears with signs of SGN degeneration do not always show a significant auditory threshold shift. Previous studies have revealed that cat cochleas with a diffuse loss of about 50 % of auditory nerves still have relatively normal thresholds as measured by behavioral tests (Schuknecht & Woellner, 1955). The amplitude of gross evoked auditory nerve responses depends on a large number of auditory nerves firing synchronously in response to sound. The loss and dysfunction of SGNs and their processes are better identified by auditory suprathreshold measurements, such as the amplitude input/output (I/O) functions of compound action potentials (CAP) (Hellstrom & Schmiedt, 1990; Kujawa & Liberman, 2009). Dysfunction of the auditory nerve can be characterized by threshold elevations, shallow slopes of I/O functions, and diminished maximum amplitudes as compared to healthy ears (see Figs. 8.4 and 8.8).
8.2.1 The Evidence of Secondary SGN Degeneration Following Hair Cell Loss
Degeneration of SGNs can occur as a secondary consequence of cochlear injury. Loss of sensory hair cells leads to a retrograde degeneration and results in a secondary SGN degeneration, a process seen in numerous animal models (see review by Spoendlin, 1984). Direct evidence is still needed to determine the primary or secondary nature of specific neuronal pathological alterations. However, there exists cumulative indirect evidence suggesting that SGN degeneration can occur after hair cell loss, including: (1) temporal patterns: loss of IHCs occurs rapidly after cochlear injury, whereas death of SGNs occurs after hair cell loss (Dupont et al., 1993; McFadden et al., 2004); (2) spatial patterns: the location of SGN loss along the cochlear spiral correlates with the location of hair cell loss (Liberman & Kiang, 1978; Bohne & Harding, 2000); and (3) manipulability of the degeneration: cochlear perfusion of the specific neurotrophic factors that are normally provided by sensory hair cell and/or supporting cells can prolong SGN survival (Ernfors et al., 1996; Altschuler et al., 1999; Stankovic et al., 2004).
Contributing factors to the degeneration of SGN after hair cell loss may include a loss of neural activity and the absence of nerve growth factors, which are critical for neuronal survival (Leake et al., 1999; Fritzsch et al., 1997; Green, 2000). In contrast to a rapid loss of hair cells in many injury models, the secondary degeneration of SGNs is often seen as a slow process with diffuse neuronal cell death. The temporal pattern of SGN death also differs across species; for example, in the rat, a loss of 90 % of the SGNs required approximately 3 months (Bichler et al., 1983). However, in the guinea pig, half of the population of SGNs was still present a year after hair cell loss (Webster & Webster, 1981). In the cat, diffuse neuronal cell loss occurred over several years (Leake & Hradek, 1988). Finally, analysis of human temporal bones suggests that SGNs can survive several decades in human ears devoid of hair cells (Nadol, 1997).
Similar to other neuronal cells in the nervous system, SGN death after hair cell loss occurs through both necrosis and apoptosis (see review by Hutchins & Barger, 1998); however, apoptosis may be the key mechanism of SGN degeneration in the cochlea. Degeneration of SGNs after hair cell death may occur through two phases. Early-phase cell death occurs as necrosis and/or apoptosis in SGNs following a loss of neural activity due to sensory hair cell loss; the later phase of degeneration results in apoptosis due to pro-apoptotic signaling caused by a chronic stress condition (e.g., loss of neurotrophic support from hair cells and/or supporting cells) (reviews by Fritzsch et al., 2004; Green et al., 2008). Using intracochlear perfusion with aminoglycoside antibiotics, Dodson showed that SGN apoptosis occurred in guinea pig cochleae after hair cell loss (1997). In that study, kanamycin sulfate or gentamicin perfusion led to a rapid loss of hair cells within 3 days and 90 % SGN death within 10 days. Many of these SGNs degenerated through the process of apoptosis, as indicated by characteristic morphological changes including condensed cytoplasm, wrinkling of the nuclear membrane, nonmarginal clumping of nuclear chromatin, and shrinkage and fragmentation of the nucleus and cytoplasm into apoptotic bodies (Fig. 8.2). There is also evidence for necrotic death in SGNs present at an early survival time after kanamycin sulfate or gentamicin perfusion (Dodson, 1997).
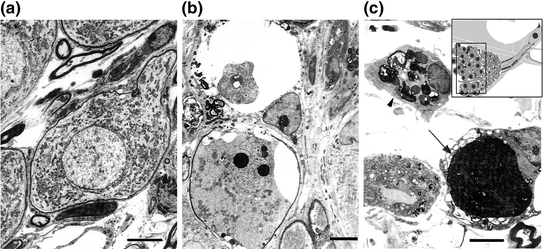
Fig. 8.2
Apoptotic cell death in type I SGNs after cochlear perfusion with aminoglycosides. a The ultrastructural features of type I SGNs from the basal turn of a normal guinea pig. b A condensed and fragmented nucleus is present in a type I SGN 10 days after aminoglycoside antibiotics were perfused into the perilymph. Separation of organelles is seen in the cytoplasm of the apoptotic neuron with a homogeneous and vesiculated appearance. c Another apoptotic neuron (arrow) is indicated by the dark chromatin masses. The arrowhead points to an activated macrophage identified by the irregular nucleus with clumped chromatin and myelin-associated cellular debris. Scale bars = 5 µm
The molecular mechanisms of SGN apoptosis have been elucidated mainly through the examination of cultured SGNs with genetic manipulation and pharmacological procedures. These in vitro studies have revealed several pro-survival signaling pathways that are involved in SGN death as a result of the absence of neural activity or the loss of neurotrophic support (see reviews by Roehm & Hansen, 2005; Green et al., 2008). These signaling pathways include, but are not limited to (1) the cyclic AMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase II and IV systems; (2) pathways involving protein kinase C (PKC), Ca2+ signaling, and mitogen-activated protein kinases (MAPK)/extracellular signal-regulated kinases (ERK) activation; and (3) the c-Jun N-terminal kinase (JNK) cell death pathway (Green, 2000; Hansen et al., 2003). In addition, recent in vivo studies have demonstrated that supporting cells in the IHC region and neuregulin–erbB receptor signaling are important for survival of adult SGN (Stankovic et al., 2004; Sugawara et al., 2005, 2007).
8.2.1.1 Primary SGN Degeneration
Animal models of sensorineural hearing loss caused by exposure to noise and ototoxic agents have been established and well-characterized morphologically and functionally for several decades. In many of these models a rapid and robust loss of hair cells was seen before a significant loss of SGNs. However, in a study of aged rat ears, Keithley and Feldman (1982) reported that neuronal degeneration exceeded IHC loss, supporting the hypothesis that neuronal degeneration is not simply retrograde degeneration after loss of IHCs, but is an intrinsic degenerative process. Primary degeneration of SGNs was also seen in aged human cochlea without a robust loss of sensory hair cells (Schuknecht & Gacek, 1993; Makary et al., 2011). In addition, primary neural degeneration was reported in some cases of noise trauma (Spoendlin, 1971; Liberman & Mulroy, 1982), aminoglycoside ototoxicity (Sone et al., 1998), and in the cochleas of white cats with hereditary deafness (Pujol et al., 1977), suggesting SGN degeneration is not a unique secondary event. A series of previous studies have found that degeneration of afferent synapses and progressive loss of SGNs occur in the cochlea when the sensory hair cells are still intact and functional after exposure to an octave-band noise at moderate levels (Kujawa & Liberman, 2006, 2009; Lin et al., 2011; see Fig. 8.4). These data strongly support that SGN degeneration can be independent of the loss of sensory hair cells.
8.2.1.2 Primary SGN Degeneration as a Result of Glutamate Excitotoxicity
Glutamate is the most common excitatory neurotransmitter in the central nervous system and is believed to play an important role in cochlear mechano-neural transduction (Bird et al., 1978; Fuchs et al., 2003). The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type glutamate receptor has been identified in type I SGNs (Pujol et al., 1985; Liberman et al., 2011). The excessive release of glutamate results in neuronal damage through excitotoxicity. The pathophysiology of excitotoxicity includes overactivation of glutamate receptors, influx of high levels of calcium ions (Ca2+) to the postsynaptic cells, and neuronal cell death (Hutchins & Barger, 1998; Martin et al., 1998).
It has been hypothesized that primary SGN degeneration occurs by means of excitotoxic neural damage. Pathological characteristics of primary SGN degeneration include massive swelling of afferent nerve terminals under the basal pole of IHCs and a total disruption of the postsynaptic membrane (Fig. 8.3; Robertson, 1983; Puel et al., 1998). The pathological alterations of afferent dendrites may be caused by excessive presynaptic release of the neurotransmitter glutamate after acoustic stimulation (Eybalin, 1993; Puel et al., 1998; Hakuba et al., 2000). Local application of glutamate agonists can induce pathologic changes in afferent dendrites similar to those induced by noise trauma (Pujol et al., 1985; Zheng et al., 1997) and the glutamate antagonist kynurenate can protect SGN dendrites from acoustic damage (Puel et al., 1998). Afferent terminals can fully or partially recover from excitotoxic damage and this recovery may play a role in the phenomenon of temporary threshold shift (TTS) (Liberman & Mulroy, 1982; Robertson, 1983; Puel et al., 1996). However, loss of presysnaptic ribbons and progressive SGN loss after TTS has also been reported in recent studies (Kujawa & Liberman, 2006, 2009; Fig. 8.4).
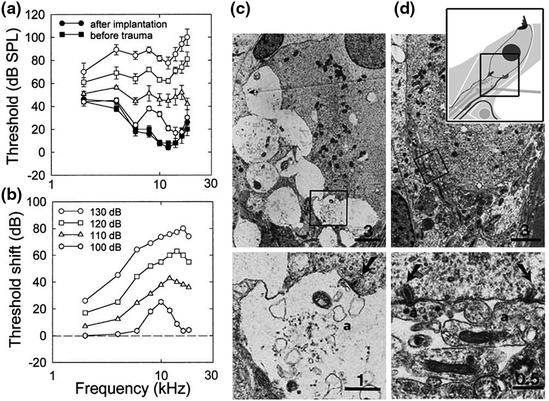
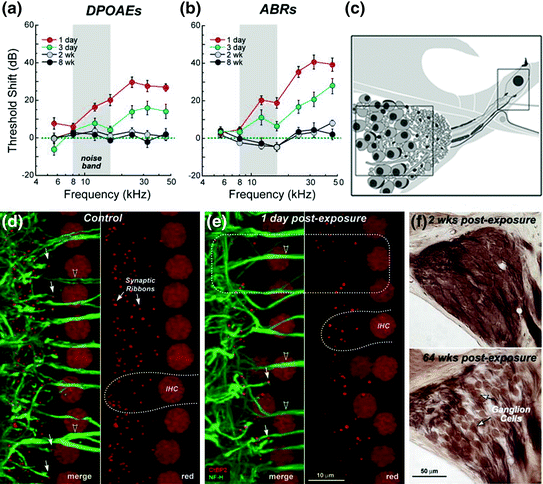
Fig. 8.3
Primary degeneration of afferent dendrites under IHCs after noise exposure in guinea pigs. a, b Compound action potential (CAP) threshold shifts are present 20 min after different levels of noise exposure (100–130 dB SPL for 15 min). Lesions in the afferent dendrites were present with noise exposures of 120 and 130 dB SPL, but not 100 and 110 dB SPL. c Pathological alterations of afferent dendrites are seen under IHCs. The preparation for transmission electron microscopic examination was processed 20 min after 130 dB SPL sound exposure. Massive swellings of afferent dendrites are present under an IHC. Bottom left panel enlargement of the area framed in (c). A presynaptic ribbon is seen at the basal pole of IHCs adjacent to postsynaptic membrane. d Significant protective effect of perilymph perfusion with kynurenate, a glutamate antagonist. No pathologic change of afferent dendrites is observed below an IHC. Bottom right panel higher magnification of the area framed in (d). An inset in the upper right panel is a schematic diagram showing the locations of images (c, d). Scale bar = 0.5 µm (Figure was modified from Puel et al., 1998)
Fig. 8.4
Primary SGN degeneration following noise-induced temporal threshold shifts. a, b Temporal threshold shifts are seen in the measures of ABRs and DPOAEs in mice after exposure to an 8–16 kHz octave-band noise at 100 dB SPL for 2 h. c Schematic representation of the locations of the SGNs and the afferent terminals under IHCs examined in the studies. d, e A rapid and robust loss of afferent synaptic ribbons (anti-CtBP2, red; arrows) in the flat preparations of cochleae at the 32 kHz region occurred 1 day after noise exposure. Auditory nerve and afferent dendrites were stained with anti-heavy neurofilament antibody (green, arrowheads). White dashed lines indicate the outlines of IHCs in the control (d) and noise-exposed (e) ears. A dashed box in (e) shows the region with significant reduction of both CtBP2+ presynaptic ribbons and neurofilament+ postsynaptic terminals. Note that the anti- CtBP2 antibody also stains IHC nuclei and anti-neurofilament also stains efferent processes to OHCs. f Cross sections show a diffuse SGN loss occurring 64 weeks after noise exposure in the 32-kHz regions of the cochlea (Figure was modified from Kujawa & Liberman, 2009)
8.2.1.3 Primary SGN Degeneration After Noise Exposure
Primary degeneration of SGNs after noise exposure has been understood largely based on experiments associated with glutamate receptor antagonists. However, questions remain on several critical issues. First, high-level noise exposures (e.g., 130 dB SPL pure tone used by Puel et al., 1998) were often applied to generate at least two types of cochlear lesions: (1) loss of hair cells starting with OHCs at lower levels then including IHCs at higher levels; and (2) a massive destruction of afferent terminals below IHCs as shown in Fig. 8.3. Although pathological nerve alterations were seen as early as 20 min after sound exposure (suggesting this pathology is independent of hair cell loss), direct evidence of primary degeneration is still needed from a model with only auditory nerve injury. Also, an evaluation of whether excitotoxic lesions in the afferent nerve terminals are able to recover fully and whether auditory nerves regenerate after noise trauma is still needed. Until recently, most of the morphological observations in these studies were performed at the ultrastructural level. Longitudinal evaluations of dynamic changes in afferent synapses and afferent nerve terminals under IHCs are extremely challenging, and quantitative analysis of morphological alterations in the auditory nerve is virtually absent from these earlier studies. Finally, because lesions are mixed in these models, comprising losses of afferent synapses, SGNs, IHCs, and OHCs, it is difficult to determine which component contributes to the various noise-induced auditory functional deficits.
Recently, newly developed genetic, biochemical, electrophysiological, and high-resolution optical approaches have provided tools for the quantitative examination of the degeneration of SGNs and their associated elements (Khimich et al., 2005; Weisz et al., 2014; Rutherford and Moser, Chap. 5). Numerous biological markers for synapses and nerve terminals have been identified and characterized, including antibodies for the presynaptic ribbon (RIBEYE/transcription factor CtBP2; Khimich et al., 2005), postsynaptic glutamate receptor patches (GluR2/3; Matsubara et al., 1996), unmyelinated nerve terminals (neurofilament; Berglund & Ryugo, 1991), and afferent terminal swellings (parvalbumin; Kujawa & Liberman, 2009). A series of studies using (1) high-powered confocal imaging of sensory epithelium, (2) three-dimensional quantification of ribbon synapse numbers, and (3) histological quantification of the neuronal cells demonstrated that a moderate level of noise exposure can cause a permanent loss of afferent synapses without hair cell damage (Kujawa & Liberman, 2009; Lin et al., 2011; Furman et al., 2013). These studies showed a rapid and selective loss of afferent synaptic ribbons under IHCs after progressive degeneration of SGNs occurring in mice after exposure to an 8–16-kHz octave-band noise at 100 dB SPL for 2 h (Fig. 8.4). Shortly after noise exposure, auditory brain stem responses (ABRs) were elevated about 40 dB concurrent with a slightly smaller threshold elevation of distortion product otoacoustic emissions (DPOAEs)—a measure of OHC function. By 2 weeks after noise exposure, ABR and DPOAE thresholds were back to normal, preexposure levels. Even though cochlear threshold sensitivity fully recovered, the ABR wave I amplitudes were reduced significantly at high stimulus levels at frequencies strongly affected by the noise. Together, these groundbreaking studies provide direct evidence that primary degeneration can occur in the inner ear in response to pathological stress conditions.
8.2.1.4 Primary SGN Degeneration and Dysfunction Associated with Gene Defects
Genetic studies of sensorineural hearing loss have progressed at a rapid pace in recent years. To date, more than 64 genes and 125 loci that link to various degrees of hearing impairment have been identified (reviews by Dror & Avraham, 2010; Angeli et al., 2012). Some of these genes play important roles in the regulation of synaptic transmission and neuronal survival and death. Deficiency of these genes likely contributes to primary SGN degeneration. Santarelli (2010) reviewed the genes associated with human auditory neuropathy, diagnostically characterized as having abnormal ABRs and completely preserved otoacoustic emissions (OAEs). Here, a brief review was given on several well-documented genes that are associated with auditory neuropathy—SLC17A8, OTOF, PJVK, and DIAPH3. In addition, animal studies of gene defects revealed two transcription factors, nuclear factor κB (NF-κB) and forkhead box O3 (FoxO3), that play important roles in maintaining the survival of SGNs and normal function of the auditory nerve and the IHC synapse (Lang et al., 2006; Gilels et al., 2013).
Vesicular glutamate transporter VGLUT3 (SLC17A8, DFNA25) and otoferlin (OTOF, DFNB9) are two key components of the afferent synapse on IHCs. VGLUT3, one of the three subtypes of vesicular glutamate transporters (VGLUT 1-3), is selectively expressed in IHCs and responsible for loading the synaptic vesicles with glutamate (Ruel et al., 2008; Seal et al., 2008). Mice lacking VGLUT3 have no auditory brainstem responses but have robust OAEs, indicating an appearance of normal OHC function. A significant reduction of IHC synapse numbers and pathological alterations of SGNs were also observed in these mice. Otoferlin is a multi-C2 domain protein essential to the exocytosis of synaptic vesicles in IHCs and the consequent action of the Ca2+ sensor triggering membrane fusion at the IHC ribbon synapse (Yasunaga et al., 1999; Roux et al., 2006). Otoferlin-deficient mice (Otof −/−) lacking exons 14 and 15, which encode most of C2C domain, are totally deaf but have preserved OAEs. Although normal IHC ribbon synapses were observed in postnatal Otof −/− mice, the pathological alterations of SGNs have not been determined (Roux et al., 2006).
Pejvakin, encoded by PJVK, is a 352-residue protein belonging to the gasdermin protein family and is expressed in cochlear hair cells, supporting cells, and SGNs (Delmaghani et al., 2006). Abnormal expression of this protein is associated with nonsyndromic auditory neuropathy DFN59 and also DFNA5, which participates in the p53-regulated cellular response to DNA damage (Masuda et al., 2006). Mice lacking pejvakin (Dfnb59t m1Ugds ) show an elevation of ABR thresholds but normal OAEs at affected frequencies. Examination of the organ of Corti via scanning electron microscope revealed no structural abnormalities, but a detailed examination of auditory nerve morphology was not included. The Diaphanous homolog 3 (DIAPH3), which encodes the diaphanous-3 protein, was mapped to the autosomal dominant auditory neuropathy, dominant 1 (AUNA1). Analysis of lymphoblastoid cells showed an upregulation of DIAPH3 mRNA expression suggesting a gain of function effect present in AUNA1-affected patients (Schoen et al., 2010). Expression of a constitutively active form of the diaphanous protein in Drosophila leads to a deficiency of auditory response from the auditory organ.
The transcription factor NF-κB has a fundamental role in regulating inflammatory responses and apoptosis in response to injury in many cell types (Barkett & Gilmore, 1999). The p50/p65 heterodimer is the predominant complex of NF κB in most mammalian cells. NF-κB is expressed and shows a low-level constitutive activity in the neurons of the central nervous system (Kaltschmidt et al., 1994). By using measures of cochlear function and histopathological evaluation, an accelerated hearing loss with correlated primary degeneration of SGNs and afferent nerve processes was seen in the SGNs of p50−/− mice (Lang et al., 2006). As shown in Fig. 8.5, marked excitotoxic-like alterations were seen at afferent terminals under IHCs of young adult p50−/− mice (1–3 month old). In contrast, no major pathological changes were seen in OHCs or the stria vascularis in the same cochleas. At 8 months of age, the density of SGNs in the basal turn of the knockouts was only about half that of wild-type mice. However, neither significant EP loss with age nor accelerated degeneration of hair cells was seen in the same cochleas, indicating that the loss of SGNs and auditory nerves is primary and independent of the degeneration of sensory hair cells.
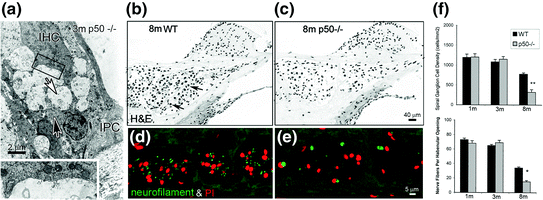
Fig. 8.5
Primary degeneration of the auditory nerve in nuclear factor-κB deficient mice. a Ultrastructural features of the basal half of an IHC and its subcellular synaptic region from the basal turn of a 3-month-old p50−/− mouse. Membranous structures presumably representing residue from degenerated cell organelles were seen in the p50−/− mice. Numerous small vesicles infiltrated with mitochondria and short profiles of cisternae appear in the cytoplasm in the base of the IHC. An inner pillar cell (IPC) and border cell enclosed the IHC and nerves consisting of intermingled afferent inner radial fibers (white arrow) and efferent spiral fibers (black arrow). The efferent inner spiral fibers and terminals (white arrow) appear normal. b–f Cross sections of the spiral ganglia in the basal turn of an 8-month-old wild type (WT) (B) and p50−/− mouse (c). g SGN counts in the basal turn in 1-, 3-, and 8-month-old WT and p50−/− mice. The density of SGNs in the 8-month-old p50−/− mice was about half that of the WT controls and the difference was significant (ANOVA, p < 0.01) (Figure was replotted from Lang et al., 2006)
FoxO3 is a transcription factor belonging to the forkhead O subclass, which is characterized by a distinct forkhead DNA-binding domain. It plays an important role in the regulation of stress response proteins in a variety of pathological conditions, including excitotoxic damage in brain tissue (Brunet et al., 1999; Davila et al., 2012). Adult mice lacking FoxO3 have elevated ABR thresholds but normal OHC function. Comprehensive histological examinations of cochlear tissues revealed that alterations of synapse locations and degeneration of the afferent nerve cause age-related hearing loss in these mice (Gilels et al., 2013).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


