Chapter 83 Eales Disease
Introduction
Eales disease is an idiopathic, occlusive perivasculitis affecting peripheral retina in young men, leading to retinal nonperfusion, new vessel formation, and recurrent vitreous hemorrhages. Eales disease was reported from the UK, USA, and Canada in the latter half of the 19th century and early 20th century.1,2 The eponym originated from the first description of a syndrome of recurrent vitreous hemorrhage in young men with epistaxis and constipation by Henry Eales.3 Most cases in the last decade have been reported from the Indian subcontinent, with a few reports from Turkey, Saudi Arabia, and Iran.4–13 However, the incidence of Eales disease appears to have declined globally, due probably to improved general health and living standards and reduced incidence of tuberculosis (TB),1,2,10 as well as to improved etiologic diagnosis in a heterogeneous group of allegedly primary vasculitides, previously amalgamated as presumed Eales disease.14 Most patients are healthy young men, aged 20–40 years; the mean age of onset is generally earlier in Asians than in Caucasians.2,15–17
Clinical features and natural history
Three sequential vascular responses which determine the natural course of Eales disease are inflammation, occlusion, and neovascularization.1,2,10,15 Most patients are asymptomatic at the stages of inflammation and occlusion. The disease starts quietly as multiple, peripheral inflammatory branch retinal vein occlusions: fine, solid white lines representing venous sheathing are the commonest clinical presentation (Fig. 83.1).1,2 As active vasculitis slowly resolves, the fuzzy vascular sheathing, with indistinct margins, becomes well defined and distinct. Retinal arteries may be involved later, but their involvement is not central to the disease presentation,1 and is generally suggestive of other conditions like systemic vasculitides.11,18 Other clinical features which suggest alternative inflammatory etiologies are exudative or focal vasculitis, cotton-wool spots,11,18 and central retinal involvement including macular and optic disc edema, choroiditis, anterior uveitis, and vitritis.2,14
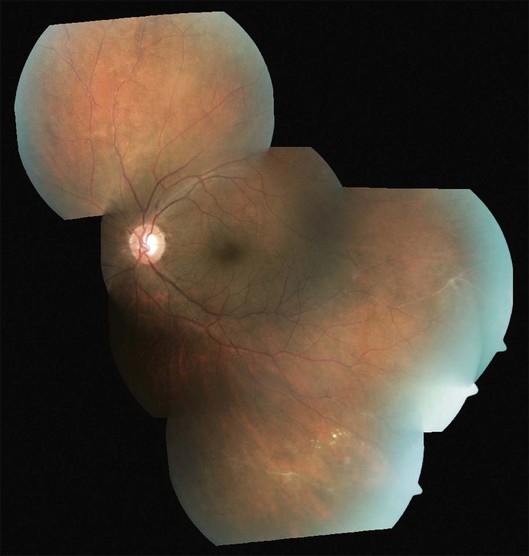
As the occlusions are primarily venous, they occur gradually, allowing development of compensatory phenomena like collaterals, microaneurysms, capillary telangiectasia, corkscrew vessels, and venous beading; some of these changes may be observed by careful examination of the apparently uninvolved fellow eye of a patient with unilateral involvement. Prolonged and extensive retinal nonperfusion eventually leads to peripheral neovascularization in up to 80% of eyes; disc neovascularization is rare.2,4,15 New vessels bleed into vitreous, resulting in the classic presentation of Eales disease: a sudden, unilateral blurring of vision or floaters15,17 (Fig. 83.2). Vision often improves, but recurrences are common. The second eye is ultimately affected in 50–90% of cases, after a gap of 3–10 years.2,4,14,15 Isolated episodes of vitreous hemorrhage usually settle down without visual deficit; recurrent bleeds lead to progressive contraction of vitreous cortex, resulting in tractional retinal detachments, secondary retinal tears, and epimacular membranes.1 Though anterior-segment neovascularization occurs in a small fraction of eyes, the prognosis is better than what is typically associated with iris neovascularization.4
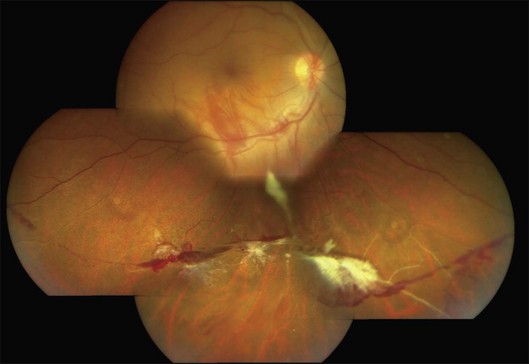
Charamis graded the evolution of Eales disease into four stages. Stage I was characterized by mild peripheral perivasculitis; stage II by extensive inflammation involving larger vessels; neovascularization and vitreous hemorrhage heralded stage III; and tractional complications marked stage IV.19 A system for grading Eales disease on the basis of the degree and extent of retinal vasculopathy, neovascular proliferations, and vitreous hemorrhage has also been proposed.20 However, the course of the disease is variable, and a fixed sequence of stages may not be followed consistently.1,2
Pathology and pathogenesis
The classic histopathological connotation of the term “vasculitis” is a type III hypersensitivity reaction with deposition of immune complexes in the vessel wall.21 This definition is not applicable to retinal vasculitis in general, and Eales disease in particular, which represents perivascular cuffing with inflammatory cells, graded by clinical appearance rather than vascular caliber or type of immune response.11,21 Most authors have therefore used the terms “vasculitis” and “perivasculitis” interchangeably in the context of Eales disease.
Central to the visually debilitating complications of Eales disease is retinal hypoxia. Inflammation causes hypoxia by an increase in the metabolic demands of cells and a reduction in metabolic substrates caused by inflammatory vascular occlusion. Hypoxia in turn triggers further inflammation, setting up a vicious cycle.22 This sequence suggest a pathogenetic role for both angiogenic factors and inflammatory cytokines in Eales disease, in a manner similar to noninflammatory vascular diseases like diabetic retinopathy. A simultaneous upregulation of vascular endothelial growth factor (VEGF), and interleukins (IL-6 and IL-9) has indeed been observed during the proliferative phases of both diabetic retinopathy and Eales disease.7 Other biochemical analyses have also implicated retinal autoimmunity, angiogenic growth factors, and oxidative stress in causing inflammation and neovascularization in Eales disease.2 Recent serologic and genetic studies have reinforced the role of cell-mediated immunity in Eales disease, particularly interleukins and tumor necrosis factor-alpha.9,12
The pathogenesis of inflammatory vascular occlusions in Eales disease remains unclear. Systemic association with neurological, vestibuloauditory, hematological, and parasitic diseases and infections have been proposed but never proven.2,15 The most frequently reported association is with systemic TB. While viable organisms have not been demonstrated from the eyes with Eales disease, polymerase chain reaction-based studies have identified mycobacterial DNA in vitreous and epiretinal tissue.23,24 These findings make the case for hypersensitivity to tuberculoprotein; the case is reinforced by the presence of Mantoux tuberculin skin test positivity in the majority of patients.2 This concept continues to be popular among current researchers.11 Several other studies have however disputed this notion by demonstrating Mantoux positivity in healthy controls, as well as Mantoux negativity among Eales patients.2,15,25
Differential diagnosis
Eales disease is a diagnosis of exclusion (see Fig. 83.3). Several ocular and systemic inflammatory and noninflammatory diseases cause retinal vascular sheathing or occlusion, which may closely resemble Eales disease. However, a battery of investigations is not necessary for every patient. A detailed history and a thorough systemic examination rule out most of the mimicking diseases; only a few tailored investigations are required to clinch the diagnosis.
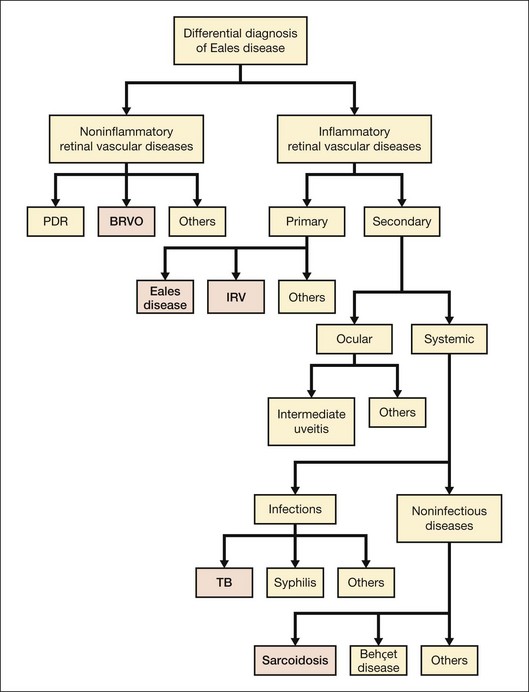
Among noninflammatory vascular occlusions, primary branch retinal vein occlusion mimics Eales most closely. The former occurs at an arteriovenous crossing; the crossing artery is frequently sclerosed. The occlusions are not multiple and peripheral like Eales, and affect older age groups. Proliferative diabetic retinopathy may also exhibit sheathing of vessels during active stage (as part of the appearance labeled “featureless retina”) or at involutional stage. Central retinal vein occlusion in a young adult should be investigated for inflammatory etiology (see diagnostic workup, below) because it represents a rare presentation of Eales.1 Coats disease, familial exudative vitreoretinopathy (FEVR) and sickle-cell disease also show similar peripheral nonperfusion and should be ruled out. Though Coats disease also occurs in male patients, they are typically younger and have unilateral disease with prominent telangiectasia, more exudation, and less neovascularization or vitreous hemorrhage. FEVR and sickle-cell retinopathy have distinctive clinical and angiographic features, as well as familial and systemic associations respectively, to distinguish them from Eales disease.
Ocular inflammatory conditions like intermediate uveitis, endophthalmitis, multifocal choroiditis, and birdshot retinopathy may have associated retinal vasculitis, but primary lesions are generally prominent and unmistakable. Systemic TB should be ruled out in every case of retinal vasculitis because of its endemic presence, especially in the Indian subcontinent.26 However, TB typically causes anterior and/or posterior uveitis along with retinal vasculitis, which is generally florid and associated with vitritis and choroiditis.26 Syphilis, like TB, needs to be screened by default because of its recent re-emergence with human immunodeficiency virus (HIV), as well as its protean and nonpathognomonic presentations, though uveitis is most frequent.27 Retinal periphlebitis occurs in about half the cases of leptospirosis, the most widespread zoonosis globally. Nongranulomatous uveitis, vitritis, and papillitis help in differentiation.28 Retinal vasculitis can also occur along with necrotizing retinitis, though arteries are involved more commonly.11
Like TB and syphilis, sarcoidosis also has no pathognomonic signs, and needs to be ruled out in most ocular inflammations. Vasculitis is typically segmental and nodular, with snowball vitreous exudates and granulomatous uveitis. Diagnosis is based on key ophthalmic signs and laboratory investigations.29 Although uncommon, Behçet disease is the classic presentation of occlusive retinal vasculitis along the Silk Route. It involves both arteries and veins, and is accompanied by severe uveitis, vitritis, and retinal infiltrates; orogenital aphthoses are diagnostic.30 Systemic vasculitides primarily cause retinal arterial occlusions rather than vasculitis: only small-vessel vasculitis, specifically systemic lupus erythematosus (SLE), is worth a mention in the differential diagnosis of Eales disease. SLE manifests more like hypertensive retinopathy than vasculitis, presenting typically with cotton-wool spots and retinal hemorrhages, and sometimes, choroidopathy.31
Primary retinal vasculitis
Once the diseases causing secondary vasculitis are ruled out, the two main conditions which remain in the picture are Eales disease and idiopathic retinal vasculitis. When the vasculitis is more posterior, sectorial and exudative; neovascularization, vitreous hemorrhage, and recurrences are uncommon; and presentation is gender-neutral, some authorities prefer the term “idiopathic retinal vasculitis” rather than Eales disease.2 Though the differentiation is rather semantic as workup remains the same (see below), Eales disease has the unusual combination of minimal peripheral vascular inflammation but extensive vascular occlusions and neovascularization leading to recurrent vitreous hemorrhages, justifying the continued use of the eponym.2
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


