CHAPTER 207 Diagnosis and Management of Tracheal Anomalies and Tracheal Stenosis
Embryology
Tracheomalacia
Tracheomalacia, which is the absence or deformity of the tracheal cartilage, can present with inspiratory and expiratory stridor, wheezing (which may be mistaken for asthma), chronic cough, and recurrent respiratory infections. It may be subdivided into two categories: primary tracheomalacia, which is an intrinsic deformity of the tracheal cartilage in the absence of any other pathology, and secondary tracheomalacia, which is due to extrinsic compression, most commonly related to cardiac or vascular anomalies, as well as tracheoesophageal fistula.1,2
Vascular Anomalies—Vascular Rings
A second but less common vascular ring arises from the persistence of the right aortic arch, with a left ligamentum arteriosum usually associated with a retroesophageal left subclavian artery, or less commonly, mirror branching of the right and left subclavian and common carotid arteries, and may present with similar symptoms as a double aortic arch.3
Vascular Anomalies—Arterial Compression
The most common cause of vascular compression of the trachea is from an anomalous innominate artery compression. A more distal origin of the innominate artery from the aorta causes compression of the right anterior trachea that endoscopically demonstrates a triangular-shaped deformity. Lifting the tip of the bronchoscope against the artery diminishes the right brachial pulse. Chest radiographs show anterior tracheal compression but no esophageal deformity.3 Often these patients present with inspiratory or biphasic stridor, cough, recurrent respiratory infections and reflex apnea, with the latter often referred to as “dying spells.”4
Pulmonary artery anomalies can also cause tracheal compression. In this situation, the aberrant left pulmonary artery arises on the right and passes between the esophagus and trachea, causing compression of the trachea and right bronchus, which is seen on endoscopy. Division of the artery with reanastomosis improves symptoms of stridor and wheezing.5
Tracheoesophageal Fistula
Tracheoesophageal fistula is a common congenital abnormality, presenting at birth with respiratory distress, excessive mucus, feeding difficulties, and aspiration. It is often associated with tracheomalacia.6 Five types have been described, with the most common abnormality occurring with the proximal esophagus ending in a blind pouch and the lower esophagus connected to the trachea via a fistula. The majority of cases are diagnosed by the inability to pass a catheter orally into the stomach.7 Severe aspiration can occur with the use of radiologic contrast studies; therefore its use is controversial. Endoscopic examination is critical in the evaluation and planning of surgical intervention.8 One half of all patients have congenital anomalies of other organ systems, including cardiac, genitourinary, and gastrointestinal abnormalities. A small percentage of patients have an intact esophageal lumen with a small connection to the trachea, known as an H-type fistula. These patients are usually less dramatically affected and diagnosis may be defied for months. Careful investigation with both contrast radiography and endoscopy is required to identify the small connection from the upper posterior trachea to the esophagus.9
Diagnosis
The severity of tracheal stenosis usually determines the age of presentation, which can be quite variable, ranging from days to weeks or even months. A long-segment stenosis with a markedly limited tracheal diameter would ordinarily present in early infancy with evidence of respiratory obstruction, especially when the airway may be compromised by secretions or infection, which produce additional incremental difficulties in adequate respiration (Fig. 207-1). Shorter segments, with a more moderate degree of obstruction in terms of tracheal diameter, may appear later in infancy or early childhood when the increasing respiratory demands of growing infants outstrip the capacity of the narrow airway to provide adequate levels of air and gas exchange (Fig. 207-2).
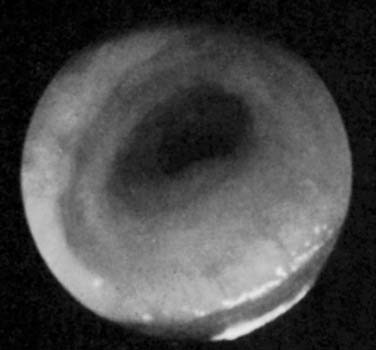
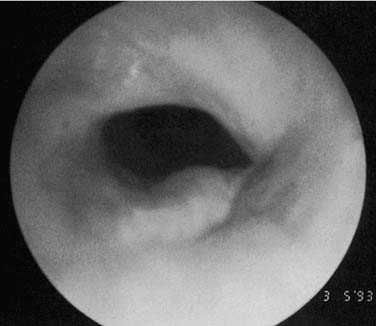
Any child who is being considered for bronchoscopy and who has atypical asthma, inspiratory and expiratory stridor, and recurrent respiratory disease should also have a potential diagnosis of tracheal stenosis. Poor weight gain and sometimes poor feeding are also associated with tracheal stenosis. Chest radiography may be helpful but is often nondiagnostic due to the dynamic aspects of the pediatric trachea. In some cases, however, a suggestion of the stenosis can be evident on close examination of the radiograph. A minority of patients with congenital tracheal stenosis present without any associated malformations and a careful assessment of particularly the cardiac vasculature is required.10
Radiology
If additional information must be obtained, contrast bronchography can be performed with a thin solution of contrast material to outline the tracheal dimensions and bronchial branchings. This contrast material must be thinned because the secretions themselves and inflammation—which is sometimes stimulated by the contrast—can cause significant problems in ventilation for the patient. Despite the inherent danger in contrast bronchography, it does provide a measurable evaluation of long-segment tracheal stenosis and any accompanying bronchial abnormalities, which are often seen in congenital stenoses. There is increasing evidence that bronchography and angiography is being supplanted by spiral CT with multiplanar reconstruction and MRI.11
Advances in CT/MRI have allowed for fine detail three-dimensional reconstructions to provide a virtual endoscopy of the airway. It is limited by not being able to provide a dynamic assessment of either tracheomalacia or bronchomalacia, may not provide enough resolution to define subtle anatomic abnormalities, and can underestimate the degree and extent of the narrowing.12 It has the advantages of being able to evaluate the airway distal to a stenosis that cannot be traversed by bronchoscopy and can be used in the measurement of the airway caliber over time. It should be considered as a complementary technique to formal rigid or flexible endoscopy in selected patients.13
Associated Anomalies
Congenital tracheal stenosis is often associated with other developmental anomalies. Cardiovascular anomalies are the most common, occurring in up to one half of cases, and include patent ductus arteriosus, ventricular septal defects, left pulmonary artery sling, aberrant subclavian artery, and double aortic arch.10 Anomalies of the bronchopulmonary, gastrointestinal, renal, or skeletal systems should also be sought. Several authors believe that the simultaneous repair of both tracheal and cardiac defects provides better postoperative management and long-term outcomes.14,15
Louksnov and colleagues thought that a key element to achieving a 97% survival rate in 37 patients who underwent surgical repair was the use of cardiopulmonary bypass, which allowed them extensive mobilization of the tracheobronchial tree and a tension-free anastomosis,14 as well as better exposure of the operative field.16
Classification
Congenital tracheal stenosis was originally classified based on anatomic morphologies as described by Cantrell and Guild,17 and was augmented by the development of a clinical symptom classification by Anton-Pacheco.11 These complementary approaches allow better correlation of anatomic findings with patient symptoms, important for surgical planning and outcome (Tables 207-1 and 207-2).
Table 207-1 Cantrell and Guild Structural Classification
| Type 1 | Generalized hypoplasia of the entire trachea |
| Type 2 | Funnel stenosis—normal proximal trachea with distal narrowing to carina |
| Type 3 | Segmental stenosis with up to three rings involved |
Data from Cantrell JR, Guild H. Congenital stenosis of the trachea. Am J Surg. 1964;108:297-305.
Table 207-2 Anton-Pacheco Functional Classification
| Type: Mild | None or occasional symptoms |
| Type: Moderate | Respiratory symptoms without respiratory compromise |
| Type: Severe | Respiratory compromise |
| Subtype A | No other associated malformations |
| Subtype B | Associated malformations |
Data from Anton-Pacheco J, et al. Patterns of management of congenital tracheal stenosis. J Pediatr Surg. 2003;38:1452-1458.
Surgery
Conservative Management
The role of conservative or nonsurgical management of congenital tracheal stenosis has recently been better defined. Historically, patients were treated conservatively because of poor surgical outcomes. There is increasing evidence that a carefully selected group of patients with congenital tracheal stenosis will resolve their difficulties with growth of the airway. This is largely due to the fact that tracheal airflow is proportional to the fourth power of the radius of the trachea. Patients with mild stenosis and occasional symptoms can be managed with respiratory physiotherapy, antibiotics in case of infection, supplemental oxygen, and close observation.11,18 Rutter and colleagues described seven patients with complete tracheal rings who did not require surgical intervention.19
Cheng and colleagues reported their experience with 22 consecutive patients, 11 who underwent surgery and 11 who were followed nonsurgically. Initial diagnosis was made with bronchoscopy and supported by radiologic and clinical findings. The patients who were observed later (1.5 vs 0.4 years), were less symptomatic, had less severe tracheal narrowing (58% vs 30% of normal tracheal diameter), and had a lower mortality than the surgical group. Six of the 11 patients in the observation group underwent serial CT measurements of the tracheal diameter. These data showed that five of six patients experienced an increased rate of tracheal growth compared with normal pediatric tracheal growth until approximately 7.5 years of age, when it then grew at a normal rate. The researchers suggested conservative management if the stenosis was not long segment and if the most stenotic segment was less than 60% of the normal trachea.18
Dilation
Dilation of strictures has traditionally been performed with Jackson or Maloney rigid dilators. This method involves serial dilation with progressively larger-diameter bougies but has a tendency to shear tissue, which may create more scarring and further stricture formation.20 In comparison, balloon tracheoplasty imparts only radial forces to the lesion and is less traumatic to the surrounding tissues. Balloon devices range from catheters developed for percutaneous transluminal angioplasty to specially designed balloons that fit over the tip of a rigid bronchoscope. Balloon-tipped catheters are connected to an inline pressure gauge and a water-filled syringe that delivers a steady, constant dilation. Serial dilation is performed with increasingly larger balloons. Balloon-tipped catheters can also be placed under radiologic guidance to confirm placement (Fig. 207-3).21
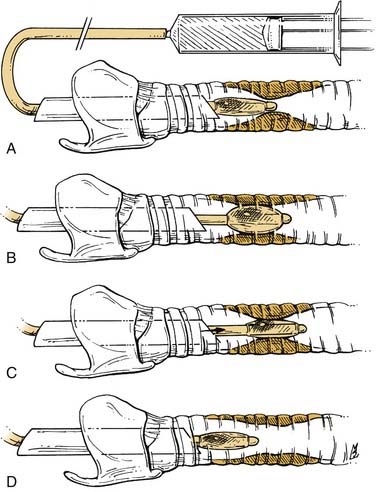
A 15-year experience of balloon tracheoplasty in 37 patients was reported by Hebra and associates.20 Short-term improvement for both congenital and acquired lesions was seen in 90% of patients, with long-term improvement in 54%. Complications were infrequent and included tracheitis, atelectasis, and pneumomediastinum. This method appears to be most useful in the management of soft granulation tissue or to aid in delaying open reconstruction in premature or medically unstable infants.
Messino and colleagues22 and Bag and associates23 have described aggressive balloon dilation of complete tracheal rings with endotracheal stenting in children younger than 2 years of age. The posterior wall of a complete ring is the weakest point, as demonstrated by their postmortem studies, and allows disruption of the trachea with subsequent fibrotic healing. This method should be reserved for critically ill patients because complications can be life threatening.
Laser Therapy
Laser therapy is often combined with dilation procedures in the endoscopic management of tracheal strictures or obstructing masses. Use of the laser allows precise tissue ablation with less trauma to the surrounding tissue compared with cryosurgery and electrocautery.24 Early reports described the use of the carbon dioxide laser with a bronchoscopic coupler and a rigid bronchoscope to manage tracheal lesions.25 With the development of improved optics and laser micromanipulators mounted directly to the microscope, visualization and precision have improved. Lesions most amenable to this technique are soft granulation and short segments of mature fibrosis. Simpson and others24 and Ossoff and associates26,27 listed the following factors as contributing to laser therapy failure: circumferential scarring, fibrosis greater than 1 cm in vertical length, tracheomalacia, and severe bacterial infection in the presence of a tracheostomy.
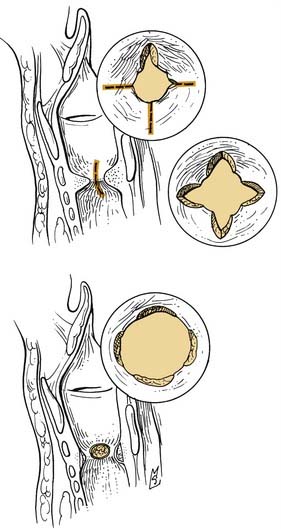
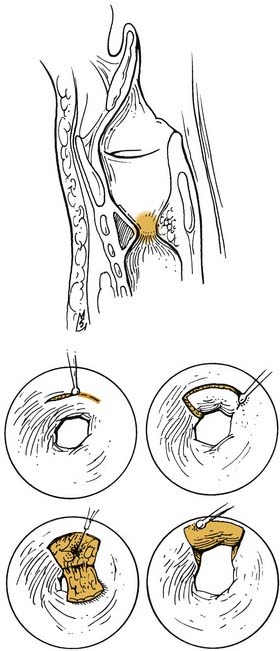
A method using radial incisions and dilation with a rigid bronchoscope for moderate to severe subglottic and tracheal stenosis was described by Shapshay and coworkers.28 Success was attributed to the preservation of islands of epithelium between the radial incisions. Results were good at 1-year follow-up (Fig. 207-4). Mucosal preservation was also advocated by Dedo and Sooy,29 who described the use of the carbon dioxide laser in the elevation of mucosal flaps. An incision is made into the superior surface of the scar, creating a trapdoor flap, and the underlying fibrous tissue is ablated with the laser with replacement of the mucosal flap (Fig. 207-5). Success with these techniques appears to be dependent on reepithelialization before scar formation, avoidance of extensive tissue damage, preservation of mucosa, and careful patient selection.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


