CHAPTER 185 Craniofacial Surgery for Congenital and Acquired Deformities
Congenital craniofacial deformities commonly occur as isolated defects and less often as part of a syndrome. The Committee on Nomenclature and Classification of Craniofacial Anomalies of the American Cleft Palate Association has organized craniofacial malformations into the following five categories: (1) facial clefts/encephaloceles and dysostoses; (2) atrophy/hypoplasia; (3) neoplasia/hyperplasia; (4)craniosynostosis; and (5) unclassified. Clinical entities such as orbital hypertelorism often exist with a syndrome that clearly fits into one of the aforementioned classifications.1 Hypoplasia of the midface and micrognathia would be incorporated into the second category. In children, hematologic disorders and overly aggressive cerebrospinal fluid (CSF) shunting can result in secondary craniofacial disorders, which can be put into the third or fifth category. Acquired deformities of the craniofacial complex also include those inflicted by means of a traumatic event. Neoplasm (third category) and its treatment are classified as acquired deformities.
Epidemiology
Craniofacial anomalies have been reported to constitute approximately one third of all congenital defects. The incidence of assorted individual deformities and syndromes varies. However, the overall incidence is considered to be 0.2 to 0.5 per 1000 births.2 Interestingly, some craniofacial deformities occur at a uniform rate across racial and ethnic populations, whereas others occur at a frequency that varies as a function of race and ethnicity.
Several craniofacial malformations appear to have a genetic etiology. Several familial types have been well documented, and transmission can occur via autosomal dominant or recessive modes. With occasional exceptions males and females tend to have similar incidences for most anomalies. Craniofacial anomalies have been reported to occur as part of a genetic condition in about 20% of cases.2 However, with the discovery of new syndromes and the recognition that “isolated defects” often have a genetic etiology, the 20% figure is probably an underestimate. Furthermore, as discussed here, the pathogenesis of craniofacial anomalies is complex, and generally it is most accurate to describe their etiology as multifactorial.
Mechanisms of Postnatal Facial Growth
It was once thought that craniofacial growth (like all other growth) was regulated, to a large extent, by the central nervous system. It is now understood to be much more complex. On a cellular level, the signal-sensitive membranes of the many types of cells respond to several different cues. On a developmental level, the functional needs of a particular structure and its relationship to adjoining structures strongly influence patterns of growth. Craniofacial bony growth is secondary to neural tissue growth and expansion. The brain as an organ mass develops early in embryogenesis. The developing brain highly influences the morphology of the basicranium, which in turn functions as a template for subsequent facial growth. The face can be viewed as a series of stratified vertical levels or fields that are strongly affected by growth of the brain and basicranium.3
Physiologically, there are two primary forces at play during the growth of individual craniofacial bones and structures, displacement and remodeling (Fig. 185-1). Growth does not indicate uniform enlargement of a structure; rather, it refers to a combination of displacement and remodeling. Displacement occurs when the bone is pushed away from its articulation with other bones (by the combined effects of growth of the surrounding soft tissue and growth center/sutural activity). Remodeling occurs by resorption and deposition of new bone that result in a net vector of growth. It is in this way that the correct tissue mass is achieved and that enlargement occurs proportionally and in the appropriate direction.
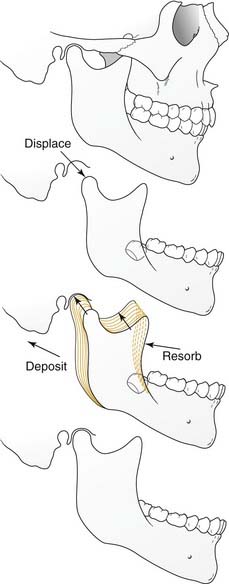
In the craniofacial complex the basicranium (along with the related soft tissues) influences the naso-orbito-maxillary complex and the ramus of the mandible, which conform through continual remodeling. Growth of the naso-orbito-maxillary complex displaces the maxillary dento-alveolus. This displacement together with growth of the mandibular ramus then affects growth of the corpus (body) of the mandible. The importance of soft tissues in growth is demonstrated in such clinical manifestations as the “adenoid face.” The entire system is plastic and in flux throughout the growing period (even after periods of growth, albeit to a far lesser extent). It is for this reason that rigid nonbiologic implants such as those of titanium have the potential to interfere with normal growth if used during active development; they lack the plasticity that is so vital to dynamic growth.4–6
Throughout the period of craniofacial growth an overriding homeostasis exists among the different fields of growth. Small aberrations in one field can be counterbalanced by compensatory growth in another, resulting in constant equilibrium. This is very important for the physician to bear in mind when planning surgical or orthodontic intervention. Treatment failure or relapse is likely if that equilibrium is significantly disrupted.7 Generally, interventions are more likely to succeed if the cause of the abnormality is addressed during periods of rapid growth. In contrast, the effect of the abnormality is more successfully treated during periods of slow growth.3
Craniofacial synostosis, or premature closure of a suture, is a good example of the morphologic consequence of defective skeletal growth. It also illustrates the importance of homeostasis in craniofacial development. The causes of craniosynostosis are likely multifactorial and the source of some controversy.8,9 It has been shown to be associated with abnormalities in sutural biology, various biomechanical forces,10 and defects in the primary growth centers of the involved bones. Sutural growth is also affected by surrounding tissue interactions. The contacting dura, for instance, influences the continued patency of sutures and helps regulate its closure.11–14
Fusion of the facial processes during embryogenesis is another fundamental aspect of facial growth that is critical to normal development. Orofacial clefting results from a defect in this process. For opposing facial processes to fuse in the midline, the contacting epithelia must be eliminated so that the underlying mesenchymal tissue can become continuous. In the palate this elimination has been shown to occur through apoptosis, epithelial to mesenchymal cell transdifferentiation, or cell migration.14–17 Underdevelopment of the mesenchyme of the involved subunits might preclude proper fusing, as can excessive apoptosis. Inadequate migration or proliferation of neural crest ectomesenchyme might have the same result. Facial clefting can also result from constricting anatomic defects. In the case of stunted mandibular growth, a Pierre Robin sequence can ensue whereby the palatal shelves are prevented from fusing in the midline.
Pathophysiology
Molecular Genetics
Several genetic defects have been identified that are directly associated with various craniofacial defects (Table 185-1). The products of these genes tend to fall into one of the following categories:
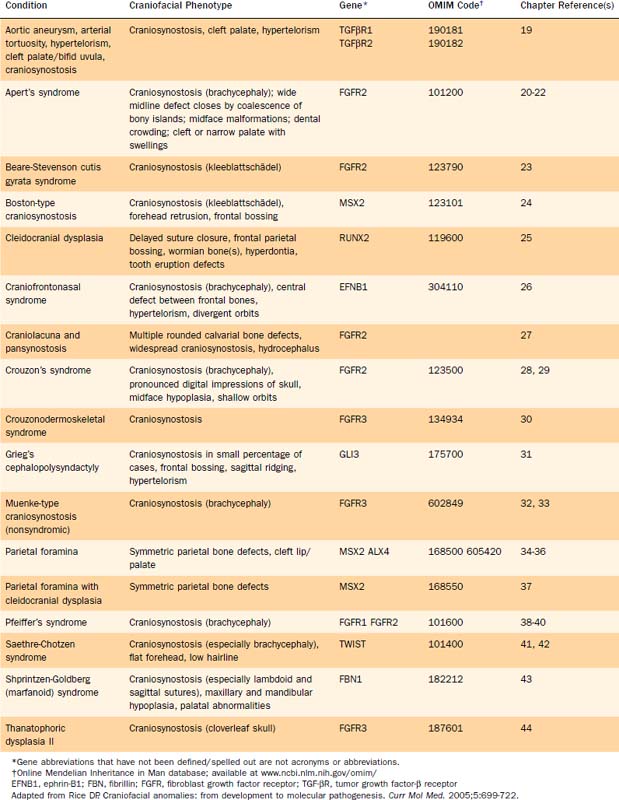
These genetic defects have been linked to syndromes as well as isolated defects. For instance, defective FGFRs have been identified as the etiologic basis of some types of craniosynostosis as well as various syndromes (see Table 185-1).18–45
FGFRs are transmembrane receptors for fibroblast growth factors (FGFs). FGFs are involved in regulating cell proliferation, differentiation, and migration through a number of pathways. The receptor is made up of an extracellular domain (receptor-ligand binding), a transmembrane domain, and an intracellular tyrosine kinase enzymatic domain (Fig. 185-2A). There are several variations of the FGFR protein—all with important cell growth functions. Signaling via FGFR1, for instance, has been shown to facilitate neural crest migration, and a defective receptor has been linked to midline facial clefting in mice.45 Defects in FGFR1 have been found to cause Kallmann’s syndrome 2, which can include cleft lip and palate (see Fig. 185-2B).46 Defects in FGFR1 and FGFR2 have been linked to syndromes such as Pfeiffer’s syndrome (in which both receptors are defective) and to conditions of which craniosynostosis is a feature.38,47 Defects in FGFR2 alone have been found in Apert’s syndrome,20,48 Jackson-Weiss syndrome,28 Crouzon’s syndrome,29 and Beare-Stevenson cutis gyrata syndrome.49 A defective FGFR3 protein is responsible for Crouzonodermoskeletal syndrome (Crouzon’s syndrome with acanthosis nigricans),30 for Muenke-type craniosynostosis,32 and also for the disease process seen in achondroplasia.50 In this disease the epiphyseal growth plates in long bones prematurely fuse. There are gross and histological similarities of long bone growth plates to the synchondroses of the calvaria. This work has led to the understanding that FGFR3 is involved in regulation of skeletal growth. In FGFR3 knockout mice (mice in which the gene that encodes this protein has been eliminated), there is severe retardation of skeletal maturation.48,51–53
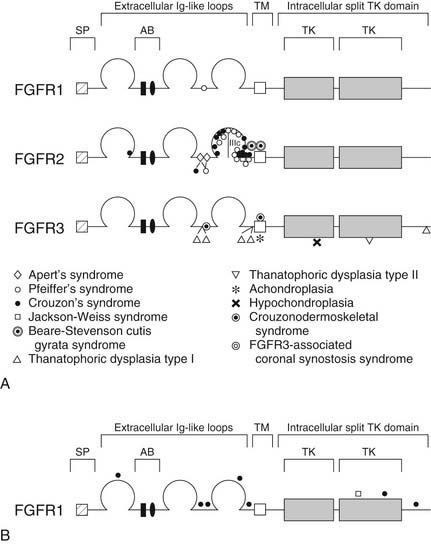
The link between known syndromes or anomalies and genetic defects, however, can be complex and difficult to characterize. Defects in each of the three domains (in different FGFRs) have been discovered in association with craniofacial abnormalities, yet defects in the same domain of one FGFR type can cause two separate syndromes. For instance, identical mutations have been discovered in patients with Crouzon’s, Pfeiffer’s, and Jackson-Weiss syndromes, suggesting the involvement of other factors in the ultimate phenotypic expression.38 Conversely, as is evident in Figure 185-2B, mutations in several distinct domains on one particular FGFR can result in the same phenotype.
As previously mentioned, defects in transcription factors and connective tissue proteins are also implicated in the pathogenesis of craniosynostosis. MSX2 is a homeobox gene that encodes a DNA binding transcription factor. It has been associated with nonsyndromic craniosynostosis (Boston type),24,54 and ALX4, another homeobox gene, has been linked to parietal foramina.34 Most cases of Saethre-Chotzen syndrome are caused by haplo-insufficiency of the TWIST gene, which appears to encode a transcription factor.41 The GLI-3 gene and the transcription factor that it encodes are defective in Grieg’s cephalopolysyndactyly (a rare syndrome that can include craniosynostosis).31,55
Genetic defects encoding several different extracellular matrix constituents can lead to anomalies of the craniofacial complex. Defective collagen, such as that seen in osteogenesis imperfecta, is clearly associated with bony abnormalities, although this disease only occasionally affects the craniofacial skeleton. Defective fibrillin (encoded by FBN1) results in Shprintzen-Goldberg syndrome with craniosynostosis and maxillary/mandibular hypoplasia.43 The defective genes responsible for Stickler’s syndrome are COL2A1 and COL11A2, which encode types II and XI collagen, respectively.56,57
Metabolic Disorders
Several metabolic derangements are known to interfere with craniofacial development. The best known is the group of disorders known as the mucopolysaccharidoses. This group of disorders is characterized by a deficiency of lysosomal enzymes that results in a buildup of mucopolysaccharides. Although there are several clinical entities, patients with some of the more severe variations of the disease have large and dolichocephalic (long and narrow) skulls with premature closure of the sagittal suture and poorly developed mastoids and paranasal sinuses. Mucolipidosis, hyperthyroidism, and rickets are also known causes of craniosynostosis.58,59
Intrauterine Causes
In utero environmental contributors to craniofacial maldevelopment fit into the following four general categories: teratogenic, infectious, nutritional, and mechanical. Fetal alcohol syndrome can involve a wide range of defects, including holoprosencephaly. Tobacco use during critical periods of embryogenesis has also been shown to adversely affect craniofacial development. Medications such as hydantoin, phenytoin, valproic acid, and isotretinoin (a vitamin A derivative used in the treatment of acne) can exert deleterious effects on embryogenesis, as can toluene, the environmental contaminant dioxin, and ionizing radiation.14,60,61
Viral infections during pregnancy have been shown to cause cleft lip/palate via mutations in PVRL1 and IRF6.62,63 Nutritional intake is also clearly linked to craniofacial development. Sonic hedgehog signaling, which is important in mediating parts of craniofacial development, is modulated by cholesterol. Therefore diets and medications that profoundly influence cholesterol levels can cause craniofacial malformations.64,65 The link between a diet deficient in folic acid and neural tube defects has been well documented and has led to the widespread supplemental addition of folic acid to many processed foods throughout the developed world.
Finally, intrauterine constraint can cause deformation of the craniofacial skeleton. Factors that have been associated with intrauterine constraint include breech presentation, persistent fetal lie, early pelvic engagement of the head, oligohydramnios, prima gravidity, multiple gestations, uterine malformations, amniotic bands, and defects in fetal neuromuscular development.66,67
Acquired Craniofacial Deformities
Several postnatal conditions predispose an infant to disrupted craniofacial growth. Hematologic diseases such as thalassemias, sickle cell anemia, congenital hemolytic icterus, and polycythemia vera are known to cause craniosynostosis. Hyperplasia of the bone marrow leads to bony overgrowth in the skull. This in turn can cause the calvarial sutures to fuse prematurely.68 Iatrogenic craniofacial anomalies can occur in patients who require ventricular shunts. If the shunt volumes in such patients are excessive, there is a lack of tension across the sutures, which produces an environment that mechanistically resembles the one seen in microcephaly. In these conditions there is an increased risk of secondary craniosynostosis.58 Etiologically, trauma and neoplasm can cause acquired craniofacial deformity, although they do so rarely.
Clinical Manifestations
Congenital Craniofacial Abnormalities
Craniofacial anomalies range from mild functionally asymptomatic defects (certain single-suture craniosynostoses) to severe anomalies that are not compatible with life (forms of holoprosencephaly). Typically, the pediatrician consults the craniofacial surgeon about a neonate who has an abnormally shaped head or unusual appearance. A fundamentally important point is to suspect the deformity to be part of a wider syndrome and to initiate a comprehensive examination in search of other abnormalities. However, the majority of cases of craniosynostosis are isolated with no associated anomalies.69 Descriptions of some of the more common craniofacial abnormalities follow.
Craniosynostosis
Craniosynostosis refers to premature closure of the involved bony suture. The consequence is that growth is inhibited in the direction perpendicular to the suture line (Fig. 185-3). Ensuing developmental growth occurs in a compensatory pattern that permits, to the extent possible, continued expansion of the intracranial content in unrestricted directions. It must be stressed that the compensatory growth that occurs is the normal biologic response to sutural constraint. The functional and aesthetic imbalance that occurs is, in part, a byproduct of normal physiology. The various types of craniosynostosis are as follows.
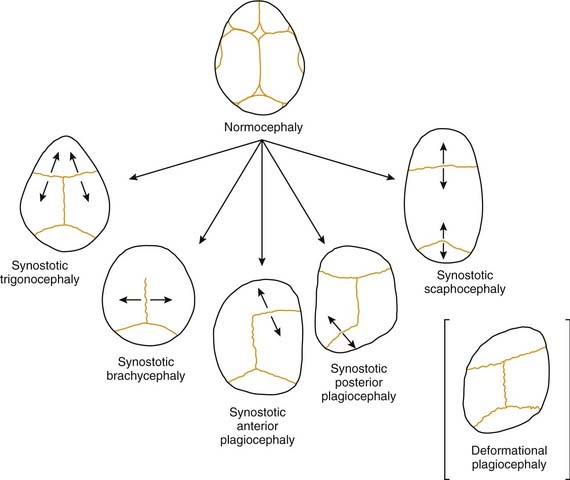
Scaphocephaly
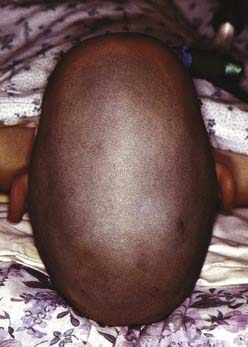
Scaphocephaly is the most common of cranial synostotic conditions. The sagittal suture, or part of it, prematurely fuses (Fig. 185-4). This anomaly prevents calvarial growth in the transverse dimension. The compensatory growth occurs in the anteroposterior (AP) direction, leaving the patient with decreased biparietal and bitemporal dimensions. The resulting head shape is long and narrow. There are several variations (“saddle deformity,” “football deformity”), depending on the extent of premature sagittal fusion and also on whether the metopic suture is involved.
Trigonocephaly
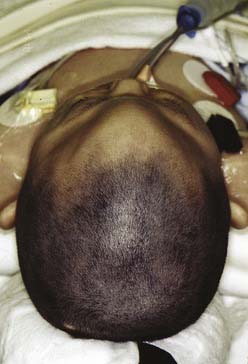
Trigonocephaly refers to a triangular forehead (as viewed from above) and is associated with metopic suture synostosis. There is a decrease in bifrontal diameter. It is a mild form of craniosynostosis with rare functional consequences (Fig. 185-5). The suture area is thickened, resulting in an anterior midsagittal ridge. This appearance has been described as resembling the keel of a ship. There can be associated hypotelorism. In mild cases there may be only suture ridging without significant forehead deformity.
Plagiocephaly
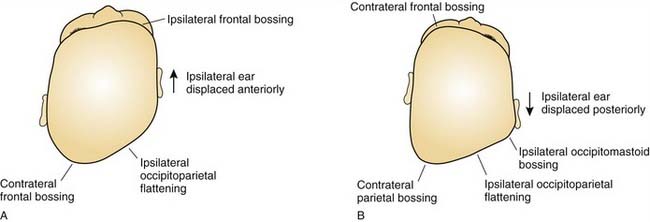
In plagiocephaly, the head is asymmetric, flattened in one or more regions. It can be either synostotic or deformational and can be anterior or posterior. Synostotic plagiocephaly refers to either unilateral coronal (Fig. 185-6) or lambdoid synostosis. In coronal synostosis the forehead of the affected side is flat whereas the supraorbital rim is elevated and displaced posteriorly. The rim lies at or posterior to the plane of the cornea although it should normally be about 1 cm anterior to the cornea. The nasal root can be deviated to the side of the defect. The contralateral forehead is bossed, and the supraorbital rim can be pushed down. The ipsilateral auricle is anteriorly displaced but the occiput is minimally affected.
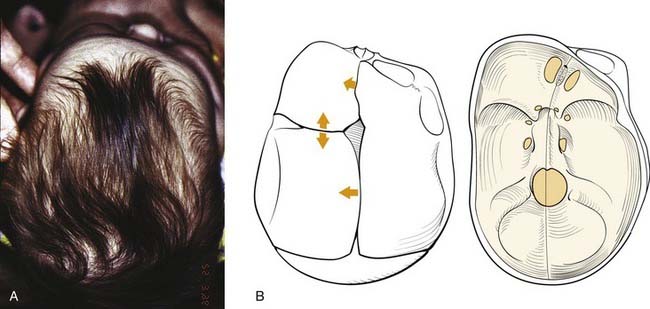
Deformational plagiocephaly is far more common and can be influenced by the previously noted intrauterine constraint factors, hypotonia, and prematurity. It is further compounded by sleeping in the supine positioning, with a predilection of the infant to sleep on one side owing to a prominent occiput and poor head control. This phenomenon has increased since the American Academy of Pediatrics, in an effort to reduce sudden infant death syndrome (SIDS), issued its recommendations to place sleeping infants in the supine position.58,69 Additionally, persistent fetal lie, vertebral anomalies, muscular disorders, ocular abnormalities as well as assorted other conditions, can cause torticollis, further aggravating sleep position problems. The sutures in deformational plagiocephaly remain open so there is no ridging. The differences in head shape between synostotic and deformational plagiocephaly are illustrated in Figure 185-7.
Brachycephaly
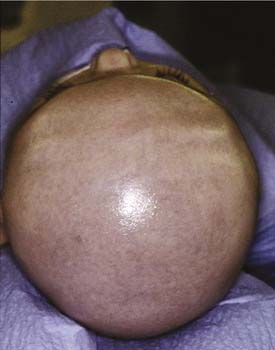
Brachycephaly is the term used for bilateral coronal synostosis (Fig. 185-8). It is often accompanied by turricephaly (tall head) or acrocephaly (pointed head). As a result of this sutural synostosis, the skull is unable to expand in the AP dimension. In order to accommodate the expanding intracranial content, the skull grows in the lateral and superior dimensions. The neonate’s cranium, therefore, has a smaller AP distance and greater lateral dimensions. The forehead comes right off the nose with a low hairline, and the glabellar depression is absent.
Acrocephaly
Acrocephaly (also known as oxycephaly and turricephaly) consists of a skull that is high and conical (Fig. 185-9). This abnormality can develop as a result of progressive postnatal involvement of both the sagittal and coronal sutures. Compensatory growth occurs through the anterior fontanel.
Kleeblattschädel
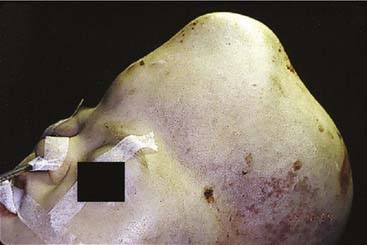
Kleeblattschädel is also known as cloverleaf skull (Fig. 185-10). It results from synostosis of all the sutures previously discussed except for the metopic suture. The bone overlying the sutures is very thick but the intervening bone is thin. The expanding intracranial content tends to cause a ballooning of the thin bone between the sutures. This compensatory growth, in addition to expansion through the remaining open sutures, gives the characteristic lobular appearance of a cloverleaf. The skull growth falls drastically short of the needs of intracranial expansion, giving rise to neurologic impairment, increased intracranial pressure (ICP), and, often, secondary hydrocephalus. These patients exhibit exorbitism and therefore are at risk for globe exposure, and their optic nerves can also be at risk for damage. This anomaly requires prompt surgical attention to release the binding skull.
Encephalocele
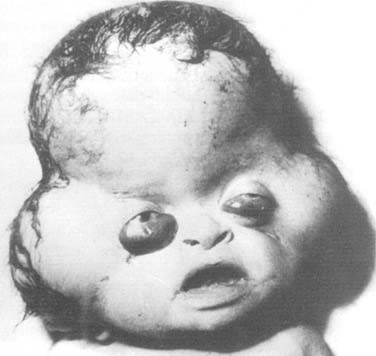
An encephalocele is a protrusion of intracranial content through a defect in the craniofacial skeleton (Fig. 185-11). It is classified according to its position in the skull. If the protruding content contains meninges, it is referred to as a meningocele. If it contains meninges and brain, it is called a meningoencephalocele, and if the protrusion additionally contains ventricle, it is known as a meningoencephalocystocele.70
Syndromes
Apert’s Syndrome
Apert’s syndrome resembles Crouzon’s syndrome in several ways. Most cases of Apert’s syndrome arise sporadically through new mutations, although some familial cases with autosomal dominant transmission have been reported.71 The disorder is characterized by brachycephaly (with resultant turricephaly) and midfacial hypoplasia (with associated orbital and dental issues). Unlike patients affected with Crouzon’s syndrome, patients with Apert’s syndrome have symmetric syndactyly of the hands and feet as well as other axial skeletal abnormalities. The palate frequently has a high arch and may be cleft. The defects in Apert’s syndrome are present at birth, and intelligence is often decreased.
Velocardiofacial Syndrome
Velocardiofacial syndrome is autosomal dominant with variable expressivity and penetrance and may be the most common syndrome after Down syndrome. There are numerous associated anomalies aside from the well-documented craniofacial, cardiac, and vascular malformations. The craniofacial anomaly broadly consists of a more open angle of flexion in the basicranium. This has an impact on the appearance of the midface and lower face (mandible). Clefting of the secondary palate can be overt or can take the form of an occult cleft that is apparent only on nasopharyngoscopy.72 Velocardiofacial syndrome can easily be overlooked because affected patients are often not obviously abnormal in appearance.



