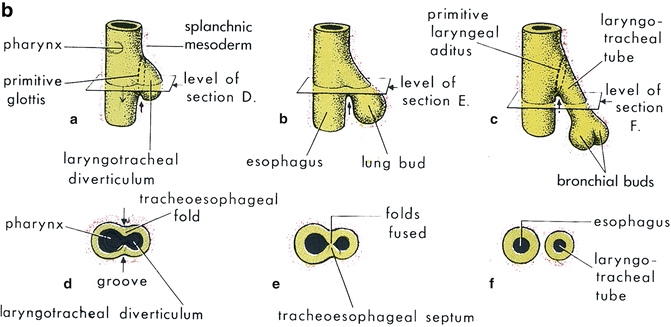
Fig. 7.1
(a, b) Schematics of embryologic development of trachea and lungs. (From Moore KL, Persaud TVN. The Developing Human: Clinically Oriented Embryology, 5th ed. Philadelphia: W.B. Saunders; 1993:228. Copyright Elsevier 1993)
Early in the fetal stage (30 mm Crown Rump [C-R] stage), the primordium of the circular muscles of the trachealis are visible as are the epiglottis, thyroid cartilage, and the hyaline tracheal cartilage rings. As the fetal stage progresses, the circular muscles of the trachea continue to develop and mature. At 100 mm CR stage both muscular layers are developed.
Anatomy of the Trachea
The trachea begins immediately caudal to the cricoid cartilage at the level of the sixth cervical vertebra (C6) and extends to the carina, where it bifurcates between thoracic vertebrae 4 and 5 (T4-5). In the adult, it measures about 11 cm and is composed of 15–20 rings of U-shaped hyaline cartilages (Fig. 7.2). In males, the trachea grows until about 20 years of age, while in females the growth halts at about 14 years of age.
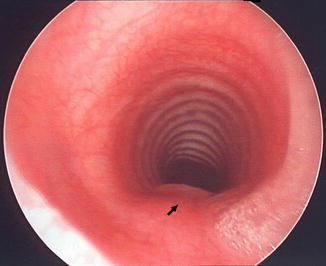
Fig. 7.2
Endoscopic photograph of normal trachea taken just above carina demonstrating horseshoe-shaped cartilages anteriorly and posterior strip of longitudinal muscle which contains no cartilage (arrow). (From Moore KL, Persaud TVN. The Developing Human: Clinically Oriented Embryology, 5th ed. Philadelphia: W.B. Saunders; 1993:227. Copyright Elsevier 1993)
The fibers of the trachea are innervated by the recurrent laryngeal nerve, and the sympathetic nerve supply is from the middle cervical ganglion. The blood supply is divided into lateral tracheal wall and lateral-anterior tracheal walls. The lateral supply is from the longitudinal vessels of the inferior thyroid artery, subclavian artery, supreme intercostal artery, internal thoracic artery, brachiocephalic trunk, and bronchial arteries at the bifurcation of the trachea. The anterior supply is via transverse segmental vessels that run in the soft tissue between the cartilages. These vessels originate in the longitudinal vessels mentioned above, and then arborize to supply the submucosal capillaries.
The venous drainage is into the left inferior thyroid vein or the laryngeal vein. The inferior thyroid veins originate on the thyroid isthmus.
Lymphatic drainage is into the pretracheal, paratracheal, and tracheobronchial nodal groups.
Tracheomalacia
Definition
This anomaly refers to laxity in the tracheal cartilage rings with resultant airway compression either due to problems related to the intrinsic strength of the cartilage or due to extrinsic compression by vasculature or masses.
History
Tracheomalacia was first described by Sprague et al. in 1933 [3] in relation to vascular compression.
Incidence
The true incidence of tracheomalacia is difficult to report as many cases of mild tracheomalacia are undiagnosed. One report estimates tracheomalacia to be present in 1 in 1,445 infants. Tracheomalacia associated with various syndromes has an incidence that parallels that of the syndrome and is, therefore, much lower [4].
Heredity
The mode of inheritance has not been defined in isolated cases. It is variable when associated with the syndromes that will be described below.
Etiology
Tracheomalacia can be a result of an intrinsic cartilaginous weakness or caused by extrinsic compression of the C-shaped tracheal rings. The intrinsic, or primary tracheomalacia, is due to a weakness and abnormality in the tracheal wall (Fig. 7.3). Secondary tracheomalacia could be due to external forces applied to the tracheal wall such as vascular compression (aberrant innominate artery, double aortic arch, or pulmonary artery sling), tracheoesophageal fistula (TEF) repair, a mediastinal mass leading to localized compression, or due to the presence of a laryngotracheal cleft [5] (Fig. 7.4). Finally, the presence of a tracheostomy tube can cause localized tracheal wall trauma and weakness (Fig. 7.5).
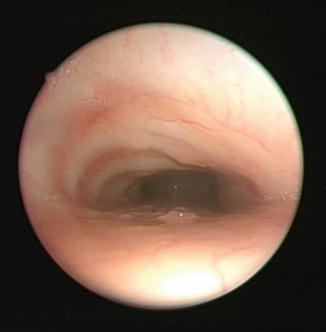
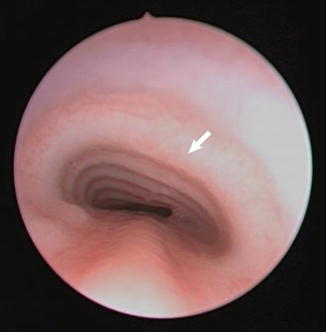
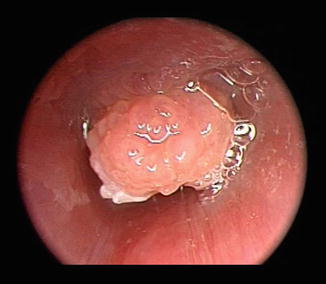
Fig. 7.3
Endoscopic photograph of trachea with intrinsic tracheomalacia. Dynamic endoscopy revealed no pulsations
Fig. 7.4
Endoscopic photograph of tracheomalacia caused by external compression from innominate artery. On dynamic endoscopy, pulsations were seen near the arrow
Fig. 7.5
Endoscopic photograph of suprastomal fibroma
Associated Malformations
As mentioned in the previous section, patients who have cardiac anomalies with vascular aberrancies and those who have had tracheoesophageal fistula repair are predisposed to tracheomalacia. The resultant extrinsic vascular compression may at times compromise the tracheal wall to such an extent that it intermittently closes off the airway causing severe cough and intermittent life-threatening events may ensue. Patients who have undergone repair of a TEF rarely (1.85 %) continue to have symptoms of tracheomalacia [6].
Clinical Features
Tracheomalacia should be considered in a child presenting with a history of expiratory stridor and/or chronic cough. The symptoms are episodic, and the child may seem healthy with no signs of airway compromise between events. In the neonate, unexplained episodes of cyanosis related to severe agitation, crying or feeding (dying spells) should lead to an investigation that includes tracheomalacia in the differential diagnosis. Cough and grunting are symptoms associated with but not exclusive of tracheomalacia. Grunting leads to an auto-positive end expiratory pressure (PEEP) that effectively stents the tracheal airway open as a compensatory mechanism. In cases of vascular anomalies, dysphagia may be a presenting feature (27 %) as well [7].
Diagnosis
The gold standard used to diagnose tracheomalacia is bronchoscopy [8]. A high index of suspicion is important, however, as tracheomalacia may be misdiagnosed as asthma especially in the older patient. Flexible and rigid bronchoscopy are employed to perform the airway evaluation. Rigid bronchoscopy provides high-definition detailed information to evaluate for laryngotracheal clefting and subglottic stenosis that can often be missed on flexible exam. However, the rigid bronchoscope can stent the trachea during the evaluation, thus under-estimating the tracheomalacia severity or missing more subtle findings. The endoscopic findings of tracheomalacia will include flattening of the tracheal rings with a “fish mouth” appearance of the distal airway (Fig. 7.3). In cases of extrinsic vascular compression, the following three characteristic findings are identified: (1) oblique flattening of the right anterior wall above carina (aberrant innominate artery) (Fig. 7.4), (2) triangular compression above the carina (double aortic arch), and (3) flattened lower tracheal wall with a collapsed right mainstem bronchus (pulmonary artery sling).
Several adjunct studies may also be helpful depending on the severity of findings or symptoms. These include barium swallow, magnetic resonance imaging or computed tomography with or without angiography to evaluate extrinsic compression/vasculature, and echocardiography in cases of suspected cardiac/vascular anomalies.
Management
In the majority of cases, in which symptoms are mild, conservative observation with parental reassurance suffices, as the disease is self-limiting. Usually, the symptoms resolve by 1–2 years of age without surgical intervention [9]. If significant tracheomalacia is identified, with worrisome symptoms including oxygen desaturations, bradycardias, and apnea, several approaches have been utilized, depending on the etiology. For intrinsic tracheomalacia, tracheostomy tube with or without continuous positive airway pressure (CPAP) is recommended. Vascular surgery for those patients with identified vascular anomalies that cause significant airway obstruction can lead to resolution of the symptoms without the need for a tracheostomy tube. Some patient will require the placement of temporary artificial airway until future growth if symptoms persist despite vascular reimplantation. Distal carinal tracheomalacia and bronchomalacia can be managed with CPAP. In some cases, airway reconstruction can remove localized segments of tracheomalacia. This option is reserved for the older child with short segments of collapse, or in cases where an indwelling tracheostomy tube led to suprastomal collapse.
Tracheal Cartilaginous Sleeve
Definition
Tracheal cartilaginous sleeve (TCS) is a congenital malformation characterized by fusion of the tracheal rings that may be isolated to a few tracheal arches, include the entire trachea, or extend beyond the carina into the bronchi. In this condition, distinct tracheal rings cannot be identified [10]. TCS has been reported only in children with craniosynostosis syndromes [11].
Incidence
No data are available on the prevalence of TCS in cartilaginous sleeve (CS) syndromes [12].
Heredity
TCSs are associated with the craniosynostosis syndromes. In a report by Hockstein et al., five of six patients with Pfeiffer syndrome had TCS [10]. All patients had mutations in the Fibroblast Growth Factor Receptor 2 (FGFR2), transmitted in an autosomal dominant pattern.
Etiology
TCSs are associated with the craniosynostosis syndromes. Pathologic evaluation of postmortem tracheal specimen showed cartilaginous sleeves with posterior interruption but lacking a normal pars membranacea. Davis et al. [12] demonstrated that the stained and cleared tracheas all demonstrate variable ring formation, usually limited to the posterolateral aspect. The functional significance of TCS, if any, is unknown. The formation of TCS implies a common mesenchymal defect in which normally discrete structures fuse and is probably analogous to other mesenchymal abnormalities seen in these patients.
Associated Malformations
TCSs are exclusively associated with the craniosynostosis syndromes and have been described in: Pfeiffer, Crouzon, and Goldenhar’s syndromes.
Clinical Features
The presence of TCS may be clinically subtle, as many craniosynostosis patients with this finding are asymptomatic until later in age. In a 1992 report by Inglis et al., four newborn patients developed recurrent lower respiratory tract infections, reactive airway disease, and chronically retained secretions attributable to this anomaly. Neither tracheal stenosis nor stridor was encountered [13]. Many of the tracheal anomalies in these patients are detected later when the patient undergoes evaluation of other upper airway anomalies such as obstructive sleep apnea from midface obstruction or from adeontonsillar hypertrophy.
Diagnosis
A high index of suspicion in patients with the craniosynostosis syndromes is of paramount importance, as TCS has only been described in this select group of patients. Bronchoscopy will delineate the degree of subglottic, tracheal, and bronchial involvement (Figs. 7.6 and 7.7). Due to the rigid nature of the trachea minimal to no respiratory movement is observed during endoscopy.
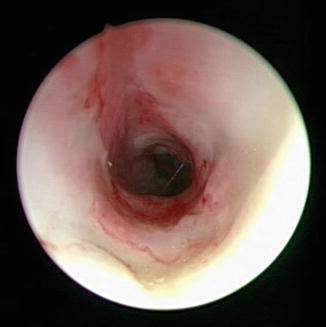
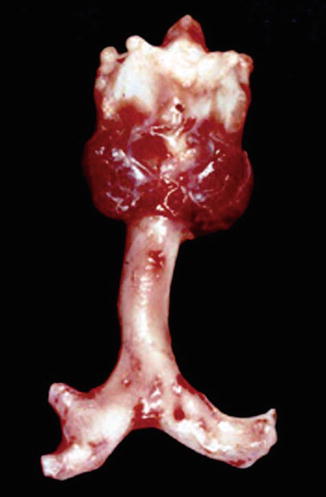
Fig. 7.6
Endoscopic view of tracheal cartilaginous sleeve that consists of a segment of fused cartilages. Note lack of discrete tracheal cartilage
Fig. 7.7
Postmortem photograph of an airway with tracheal sleeve
Management
Therapy is patient directed. In select patients with significant airway compromise or deteriorating pulmonary disease due to toilet issues, a tracheostomy is advisable and may extend the life expectancy [14]. Noorily et al. reported that 45 % of children with Crouzon’s syndrome required tracheostomy tube placement [15]. However, many of these children also had inherent obstructive sleep apnea related to the maxillofacial deformities (choanal stenosis, adenotonsillar enlargement, midface hypoplasia). Close monitoring of the airway in the tracheotomized patient is essential, as granulation tissue and tube obstruction have frequently been encountered in these patients [10]. This is thought to be related to the lack of pliability of the trachea, leading to difficulty fitting an appropriately shaped tracheostomy tube, localized trauma, and fibrosis [16]. Tracheal resection or slide tracheoplasty have been reported when less than 40 % of the length of the trachea is involved; however, these may be risky procedures due to the tendency for poor healing noted in this condition [17].
Complete Tracheal Rings
Definition
Complete tracheal rings refer to the most common congenital malformation leading to tracheal stenosis. This condition is characterized by circumferential cartilaginous tracheal ring with no posterior pars membranacea. The pars membranacea is the muscular posterior wall of the trachea which normally lacks tracheal rings.
History
Incidence
Symptomatic tracheal stenosis due to complete tracheal rings is rare and the incidence of undiagnosed complete tracheal rings is unknown [21].
Heredity
Unknown.
Etiology
Unknown.
Associated Malformations
Complete tracheal rings have been reported to be associated with an aberrant left pulmonary artery (pulmonary artery sling) in 35 % of cases [18, 22]. In a separate review, 50–60 % of patients with pulmonary artery slings had complete tracheal rings and long segment tracheal stenosis. Associated intracardiac anomalies have been reported to occur in 24 % of patients with complete tracheal rings [22]. In addition, patients with trisomy 21 have been shown to have an a higher risk of having complete tracheal rings. A recent report showed that 17.5 % of patients with complete tracheal rings (7/40) had Downs syndrome [23].
Clinical Features
Episodic or progressive respiratory distress, biphasic stridor, “washing machine” breathing sounds [24], cyanosis or a difficult intubation have all been reported as the clinical presentation of complete tracheal rings. Symptoms may often get exacerbated by an upper respiratory infection. Due to the distal trapped secretions, recurrent pulmonary infections can be seen.
Diagnosis
Bronchoscopy is the standard for diagnosis of complete tracheal rings (Fig. 7.8a, b). The endoscopy allows direct visualization of the trachea, ruling-out any other synchronous lesion of the upper airway. Other diagnostic modalities in the stable patient include magnetic resonance imaging (MRI) or computed tomography (CT) imaging to evaluate the pulmonary vasculature (Fig. 7.9a–c). An echocardiogram should be performed in all patients to rule-out pulmonary artery sling.
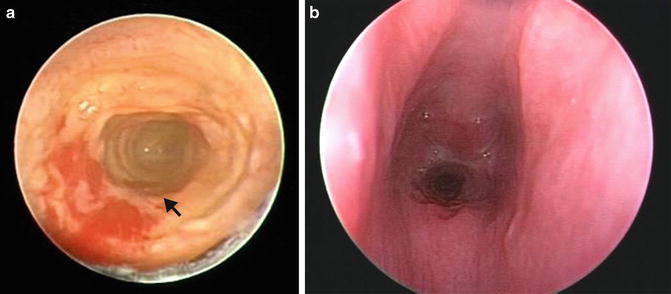
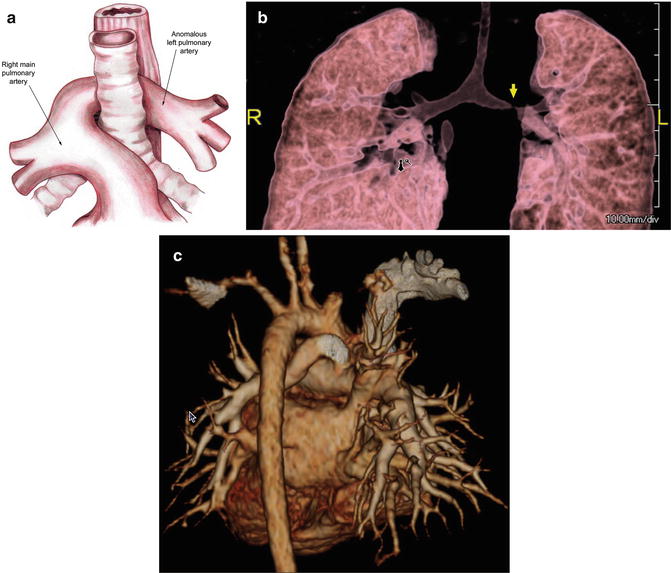
Fig. 7.8
Endoscopic view of complete tracheal ring in two patients which demonstrates (a) lack of normal pars membranacea (arrow). (b) More severe midtracheal stenosis (arrowhead)
Fig. 7.9
(a) Schematic image; (b, c) 3D rendered volume CT angiogram images demonstrating pulmonary artery sling (arrow on b)
Tracheoesophageal Fistula (TEF)
Definition
Tracheoesophageal fistula (TEF) refers to a communication between the trachea and esophagus. A TEF may be congenital or acquired.
History
The first description of a TEF dates to 1697, Thomas Gibson described a neonate with an esophageal atresia and TEF. The first surgical reconstruction of this lesion was described in 1941 and was associated with a 70 % mortality rate [25].
Incidence
Congenital esophageal atresia and/or TEF have an incidence of 1 in 3,000–4,500 live births [26]. Acquired TEFs are rare and the incidence is not known.
Heredity
VATER/VACTERL (Vertebral, Anal, Cardiac, Tracheo-Esophageal Fistula, Esophageal atresia, Renal, Radial dysplasia) association is a relatively common, nonrandom multiple-malformation condition. The incidence is reported to be 1.6 in 10,000 [27]. No consistent or recurring etiology has been established for the VATER/VACTERL association, but it has been more frequently reported in the offspring of diabetic mothers. Most cases occur as sporadic events with low recurrence risk. The pattern of malformation has generally been a sporadic occurrence; thus, there is probably a low recurrence risk (<1 %) [27].
Etiology
The trachea and esophagus both originate from the primitive foregut. During the fourth to sixth weeks of gestation, the caudal foregut forms a ventral diverticulum that later develops into the trachea. The common membrane between the esophagus and trachea, the tracheoesophageal fold, creates a septum between the two, and if it deviates posteriorly, a communication between both tubes will form. The exact cause is unknown, but it is thought to result from a developmental disorder in formation and separation of the primitive foregut into trachea and esophagus. It is generally felt to be a heterogeneous disorder with the various yet-to-be established etiologies working through a common embryological pathway [27].
Acquired TEF can result from localized trauma after ingestion of toxic foreign bodies such as a battery, localized infection which devascularizes the wall of the esophagus, malignancy or from traumatic or prolonged intubation of the esophagus or trachea (either with an endotracheal tube or a tracheostomy tube). Cuff pressures exceeding 30 cm H2O can promote trachealis necrosis and a consequent acquired TEF.
Associated Malformations
Nearly 50 % of patient with a congenital esophageal atresia/TEF have one or more anomalies, with cardiac anomalies being the most commonly recognized [26]. Other recognized associations are chromosomal abnormalities (Trisomy 21 and 18, 8 %), and gastrointestinal malformations as seen in VATER/VACTREL and CHARGE: imperforate anus, esophageal atresia, pyloric stenosis, duodenal atresia, and annular pancreas (38 %) [26]. The association between TEF, vertebral anomalies, anal atresia, and radial dysplasia was first described by Quan and Smith in 1972 [28].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


