Fig. 8.1
A frontal schematic representation of a 5-mm human embryo at the fifth week of gestation. Sagital sections taken through the branchial apparatus demonstrate the anatomic relationship of external clefts and internal pouches as well as the derivation of important head and neck structures (Reprinted from Seminars in Pediatric Surgery, vol. 15, Waldhausen JHT, “Branchial cleft and arch anomalies in children,” pp. 64–69, copyright 2006, with permission from Elsevier)
Table 8.1
Structures developed from branchial arch components
Arch | Artery | Nerve | Muscle | Skeletal | Ligament |
|---|---|---|---|---|---|
First (mandibular, Meckel’s) | Internal • Maxillary • Artery | Trigeminal | Muscles of mastication | Malleus | Anterior malleolar |
Mylohyoid | Incus | Sphenomandibular | |||
Anterior belly of digastric | |||||
Tensor tympani | |||||
Tensor veli palatine | |||||
Second (hyoid, Reichert’s) | Stapedial | Facial | Muscles of facial expression | Stapes | Stylohyoid |
Stapedius | Styloid process | ||||
Stylohoid | Lesser cornu and upper body of hyoid | ||||
Posterior belly digastric | |||||
Third | Internal and Common Carotid arteries | Glossopharyngeal | Stylopharyngeus | Greater cornu and lower body of hyoid | |
Fourth | Right: Subclavian | Superior laryngeal | Cricothyroid | Thyroid | |
Left: Aorta | Pharyngeal constrictors | Cuneiform | |||
Epiglottis | |||||
Sixth | Right: Right pulmonary artery | Recurrent laryngeal | Intrinsic muscles of larynx | Cricoid | |
Left: Left pulmonary artery, ductus arteriosus | Corniculate | ||||
Arytenoid |
In normal development, the first cleft will persist as the external auditory canal. The remaining three clefts develop into what is collectively known as the cervical sinus, which obliterates during the sixth and seventh weeks as the second arch grows inferiorly (Fig. 8.1).
The first pharyngeal pouch develops in to the middle ear and Eustachian tube, while the second pouch forms a portion of the pharynx including the palatine tonsil. The third pouch will later subdivide to become the thymus and the inferior parathyroid glands, whereas the fourth pouch derivative is the superior parathyroid gland. The fifth pouch is rudimentary and becomes part of the fourth pouch, ultimately developing into the parafollicular c-cells within the thyroid gland [3].
The first arch mesenchyme forms the majority of the malleus, incus and provides the framework for mandibular development. Muscle derivatives of the first arch correspond to those innervated by the trigeminal nerve. The first arch vascular structure becomes the internal maxillary artery. The second arch mesenchyme develops into the stapes suprastructure, styloid process, and portions of the hyoid. Muscle derivatives of the second arch correspond to those innervated by the facial nerve. The second arch artery is the stapedial, which typically obliterates but in rare cases can persist [4]. Third arch derivatives include the remainder of the hyoid bone, glossopharyngeal nerve, stylopharyngeus muscle as well as the common and internal carotid arteries. The fourth and sixth arches go on to form the framework of the larynx, its innervation and musculature. These arches also form the blood vessels within the mediastinum.
Definition
Branchial cleft anomalies can be divided into cysts, sinuses and fistulae. A cyst is an epithelial lined sac with no communication to the skin or the pharynx. A sinus has a connection to one surface (external or internal), and a fistula forms a communication between two epithelial surfaces (typically cutaneous and pharyngeal) [2].
History
Hunczowski has been credited with the first description of a cervical fistula in 1789; however, this anomaly was not attributed to the branchial apparatus until noted by Von Ascherson in 1832 [2]. Since the initial description, there have been extensive reviews in the literature regarding the embryology, diagnosis, and management of these conditions.
Heredity
Branchial cleft anomalies are most commonly sporadic but can be associated with inherited disorders such as branchio-oto-renal syndrome. This autosomal-dominant condition consists of hearing impairment, auricular anomalies, branchial cleft remnants, and renal abnormalities [5].
First Branchial Cleft Anomalies
Incidence
Etiology
Duplication or failure of obliteration of the embryologic tract is the likely etiology of these lesions. First branchial cleft anomalies are subdivided using the Work classification [8]. Type 1 anomalies are ectodermal in origin and consist of a duplication of the membranous portion of the auditory canal. They course medial to the conchal cartilage and can extend into the post-auricular crease. They pass anterior and deep to the lobule, stay superficial to the facial nerve and parallel to the normal external auditory canal. Type 2 anomalies have both ectodermal and mesodermal components. The external opening is near the angle of the mandible (Fig. 8.2), while the tract courses superiorly and can be closely associated to the facial nerve. The anomaly may present with a sinus inferior to the membranous ear canal or open into the canal itself, typically at the bony-cartilaginous junction [8].
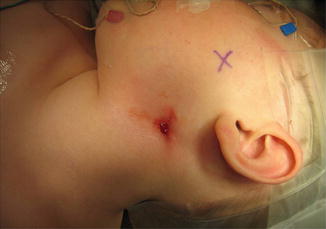
Fig. 8.2
Photo demonstrating the characteristic opening associated with a Type II, first branchial cleft anomaly
Clinical Features
These lesions can present with findings attributable to the ear, parotid or neck regions. Otologic signs include otorrhea, which can be mucoid or purulent. Parotid signs generally include inflammation and can resemble primary parotitis with rapid gland enlargement and associated tenderness. Cervical signs include cellulitis, abscess, and drainage from a pit inferior to the mandible (Fig. 8.2).
Diagnosis
First branchial cleft anomalies can be a diagnostic challenge and are often misdiagnosed and receive non-curative treatment (refer to Chap. 2). They must be differentiated from preauricular sinuses and tags, which have a different embryologic origin. A complete history and physical examination is the initial step in arriving at a diagnosis. Otoscopy should be performed to assess for any communication with the external auditory canal or attachment to the tympanic membrane. Facial nerve function should be recorded. Cervical skin should be closely examined for signs of a pit or sinus tract. Computed tomography (CT) or magnetic resonance imaging (MRI) scan can help delineate the course of the tract (Fig. 8.3).
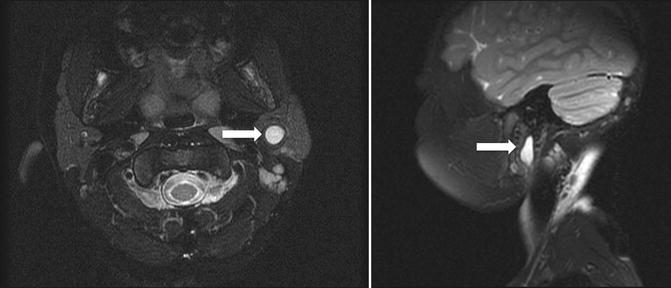
Fig. 8.3
Axial and sagital MRI images of a first branchial cleft anomaly (arrows). The lesion lies within the parotid gland and courses toward the ear canal
Management
The treatment of first branchial cleft anomalies is surgical, and complete excision is necessary to prevent recurrence. In the majority of cases, definitive surgery requires a superficial parotidectomy approach with identification and preservation of the facial nerve. The nerve’s relationship to the lesion is variable and can be medial or lateral (Figs. 8.4a, b) [9]. In the case of abscess formation, incision and drainage is often required. Purulent material should be cultured to direct antimicrobial therapy, reserving full excision for after the resolution of infection.
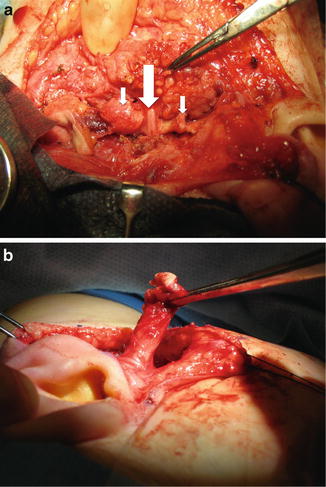
Fig. 8.4
(a) Intraoperative photograph demonstrating the close relationship between the first branchial cleft anomaly (small arrows) and the facial nerve (large arrow). The anomaly runs deep (medial) to the facial nerve. (b) Intraoperative photograph of another child with a branchial cleft anomaly that remained superficial (lateral) to the nerve, which has been dissected out from the parotid gland
Second Branchial Anomalies
Incidence
The true incidence of second branchial cleft anomalies is unknown. These lesions are much more common than other branchial anomalies, accounting for 90 % of these abnormalities. Overall, second branchial cleft anomalies are second only to thyroglossal duct cysts (TGDCs) as the most commonly diagnosed head and neck malformations.
Etiology
Failure of involution of the cervical sinus with or without connection to the pharynx is the most accepted theory for the development of second branchial anomalies. Second branchial fistulas pass between the second and third arch derivatives. Anatomically, the tract begins at the inferior aspect of the anterior border of the sternocleidomastoid (SCM) muscle, courses deep to the platysma, the stylohyoid and posterior belly of digastric muscles to the carotid sheath. It stays superficial to the glossopharyngeal nerve and travels between the internal and external carotid arteries before terminating in the tonsillar fossa (Fig. 8.5).
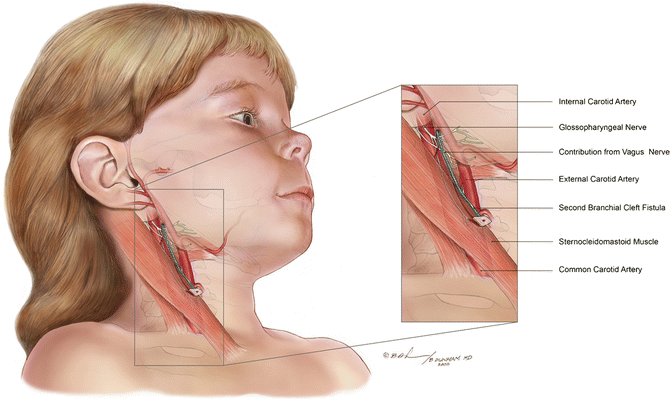
Fig. 8.5
Diagram of the right neck demonstrating the course of a second branchial cleft anomaly. The tract courses over the ninth cranial nerve (glossopharyngeal nerve) and between the bifurcation of the carotid artery (internal and external branches) to end in the inferior tonsillar fossa of the pharynx
Clinical Features
Second branchial cleft cysts often present as a soft, mobile, painless, and slow growing mass in the lateral neck. They often present after a respiratory tract infection and can become abscessed requiring incision and drainage. The opening of a sinus or fistulous tract can be identified on the anterior border of the ipsilateral SCM muscle (Fig. 8.6) and may present with intermittent non-purulent drainage.
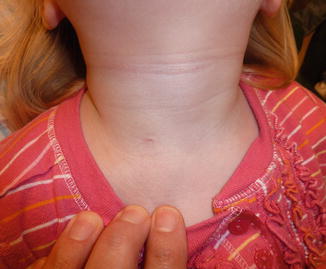
Fig. 8.6
External pit along the anterior border of the SCM muscle associated with a second branchial cleft anomaly
Diagnosis
Lateral neck swelling in the pediatric age group has a broad differential diagnosis (Table 8.2). The diagnosis can be narrowed significantly when a presence of a pit is present on the surface of the neck, although differentiation from other branchial anomalies can be challenging. An infected branchial cleft cyst needs to be differentiated from suppurative cervical adenitis. Although many imaging modalities have been used in the diagnosis of branchial cleft anomalies, CT scan remains the study of choice in most circumstances.
Table 8.2
Differential diagnosis of lateral neck mass in children
Branchial cleft anomaly |
Lymph node |
Vascular malformation |
Sternocleidomastoid tumor of infancy |
Thymic cyst |
Laryngocele |
Management
Treatment of second branchial anomalies requires complete surgical excision. When removing a cyst it is imperative that one attempts to identify and remove an associated tract if there is one present in order to minimize the risk of recurrence. For sinuses and fistulae, the tract must be completely excised. Some surgeons advocate removing the tonsil when the tract is excised to prevent recurrence within the lower tonsillar fossa. Identification and preservation of surrounding structures along the tract’s path is essential. Gentle probing of the sinus/fistula with lacrimal probes can be helpful. Methylene blue injected into the tract is another commonly used technique to allow for tract delineation.
Third and Fourth Anomalies
Incidence
Third branchial cleft anomalies are rare with just over 200 cases reported in the literature [10]. Lesions involving the fourth cleft are extremely uncommon.
Etiology
As with other branchial cleft anomalies, failure of the tract to involute is accepted as the most likely cause. The path of these lesions is based on the embryology. Third cleft lesions will travel deep to the internal and common carotid arteries as well as the glossopharyngeal nerve. The tract will then course through the thyrohyoid membrane to enter the cranial portion of the pyriform fossa. A complete fistulous tract of the fourth branchial cleft or pouch has not been reported so the course of the lesion is speculative [2]. The suspected course of these anomalies will be different depending on the side of the lesion. On the right side, the tract will loop around the subclavian artery, course deep to the internal carotid artery, ascend superficial to the hypoglossal nerve, and then descend to enter the apex of the pyriform fossa. On the left side, the lesion travels around the arch of the aorta with the remainder of the course mimicking that seen on the right.
Clinical Features
These lesions can have a similar presentation to the more common second branchial anomalies with some important variations. Both can present as a slow growing lateral neck mass and have an external pit along the anterior border of the SCM muscle, similar to second cleft anomalies. A systematic review of the literature identified that 89 % of reported third arch anomalies were on the left side and often present with abscess formation or acute suppurative thyroiditis [10]. In the setting of infection, hypoglossal nerve palsy may be present. Fourth cleft anomalies can present with neck pain, thyroid abscess, and recurrent upper respiratory tract infections [5].
Diagnosis
Diagnosis is based on history and physical exam with imaging including contrast enhanced CT, MRI, and barium swallow being most useful [10]. High resolution scans can sometimes identify the course of the tract, allowing the type to be identified, although in many cases the diagnosis will often be made intraoperatively based on the course of the tract. Because of the possible communication of the sinus or fistula into the pyriform fossa, direct laryngoscopy can aid in the diagnosis. A sinus or fistulous opening may be identified (Fig. 8.7) and in the setting of acute infection, purulent drainage can be seen in the pyriform fossa. If not infected, gentle massage of the neck can allow saliva to be expressed from the sinus.
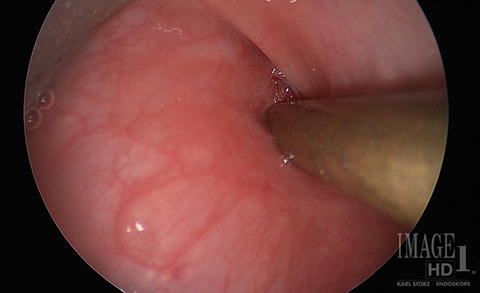
Fig. 8.7
View of the left pyriform fossa with sinus opening identified in a patient with recurrent neck abscesses. The probe is in the sinus
Management
Traditionally, the mainstay of treatment of third and forth branchial anomalies has been surgical excision. Knowledge of the relevant embryology guides the surgeon through the expected path of these lesions. Exposure of the laryngeal framework and possible removal of a cartilaginous window to access the pyriform sinus may be required. A fourth cleft anomaly requires an ipsilateral thyroidectomy to completely remove the tract [2]. Abscesses (Fig. 8.8) should be managed with incision and drainage and appropriate antimicrobial therapy, with definitive surgery delayed until resolution of inflammation. Recently, an alternative management strategy of third and forth anomalies has included identification of the fistula opening within the pyriform fossa with endoscopic cauterization (Fig. 8.9) [10, 11]. This approach has encouraging results without the potential morbidity of an open procedure.
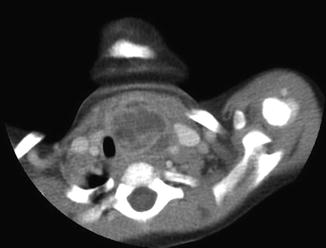
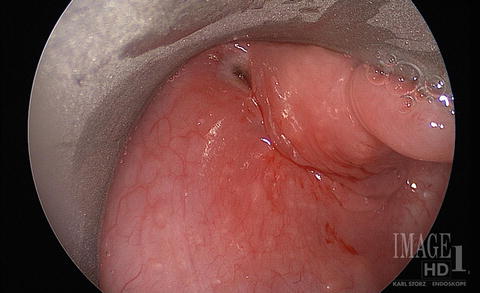
Fig. 8.8
CT scan image of a child with an abscessed third branchial cleft anomaly
Fig. 8.9
View of sinus opening in pyriform fossa after being cauterized. Note the blanching of the mucosa around the opening of the sinus
Thyroid
Embryology
The thyroid gland begins to arise during the end of the third week of gestation. It is formed by a single medial anlage and paired lateral anlages [2]. The larger median anlage arises on the floor of the primordial pharynx. It begins as an evagination termed the thyroid diverticulum and lies caudal to the developing oral tongue. The lateral anlages are derivatives from the ventral portion of the fourth branchial pouch and fuse with the median portion during the fifth week. As the embryo grows the gland passes ventral to the developing hyoid bone and reaches its final position at the level of the second and third tracheal rings [3] (Fig. 8.10).
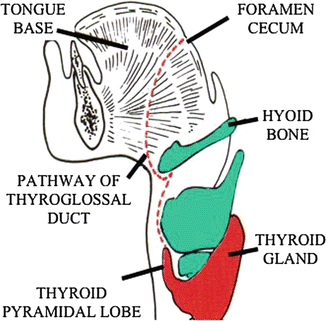
Fig. 8.10
Migratory course of the thyroglossal duct. Thyroglossal duct cysts may occur anywhere along the course of the duct if the duct fails to obliterate during development (From Moore KL, Persaud TVN. The Developing Human: Clinically Oriented Embryology, 5th ed. Philadelphia: W.B. Saunders; 1993:201. Copyright Elsevier 1993)
The thyroglossal duct is an epithelial lined tract that connects the tongue to the thyroid gland. The duct opens at the junction of the anterior two-thirds and the posterior third of the tongue at the foramen cecum. The duct typically obliterates and disappears by the fifth week of gestation. The inferior portion of the duct can often persist and is represented as the pyramidal lobe of the gland which can have fibrous stalk connected to the hyoid [2].
Definition
Thyroid tissue can be present anywhere along the duct. It can be in the form of the entire gland or small rests of cells. If the gland fails to descend, the thyroid tissue remains within the tongue base and is termed lingual thyroid. In 70–80 % of patients with this anomaly, this is the only functioning thyroid tissue present [12]. An epithelial lined cyst forming along the tract is termed a TGDC.
History
The historical aspects of TGDCs relate to their management. In 1893, Schlange first suggested resection of the hyoid bone with the cyst, which significantly reduced recurrence rates [13]. Walter E. Sistrunk is credited with developing today’s surgical treatment based on his paper published in 1920 [14]. He too discussed the importance of hyoid removal and also advocated for removal of a core of tissue above the hyoid to the foramen cecum.
Incidence
Heredity
Etiology
It is unclear what causes the thyroid not to descend normally. Failure of involution of the thyroglossal duct results in rests of cells remaining along the tract. Proliferating and secreting epithelium in response to inflammation along the tract is thought to result in a TGDC development [13].
Associated Malformations
Thyroid carcinoma can develop within TGDCs and lingual thyroid glands. The reported incidence of TGDC carcinoma is 1 % and is usually found incidentally after surgical excision [15]. This diagnosis is exceedingly rare in the pediatric age group with only 22 cases reported [20]. Lingual thyroid cancer is also rare with approximately 40 cases reported in the literature [21].
Clinical Features
A TGDC typically presents as a painless, slow growing midline neck mass. The cyst can become apparent at any age with the majority presenting before the age of five [22]. It can arise at any location along the tract, but is most commonly found below the hyoid bone. The mass is typically soft, mobile, non-tender and often moves with swallowing or protrusion of the tongue. Associated findings may include dysphagia, dysphonia, airway obstruction, and abscess formation. Although an infected TGDC may present with drainage or fistulization, it is never primarily associated with a communication to the skin because embryologically there is no communication with the surface of the neck [22]. A lingual thyroid will present as a solid mass within the posterior tongue and may be associated with feeding difficulties or airway obstruction.
Diagnosis
Although TGDC is the most common pediatric midline neck mass, there is a broad differential diagnosis (Table 8.3). History and physical exam consistent with the clinical features mentioned above are very important in the diagnosis of TGDC. An ultrasound can be useful, not only to characterize the lesion, but also to identify normal thyroid tissue within the neck and is the imaging modality of choice. Contrast enhanced CT and MRI can also be helpful, but are generally not necessary. These studies can be reserved for larger lesions, lesions presenting with a complication, or recurrent cases. Nuclear medicine studies can identify functional thyroid tissue within the lesion and the normal gland. If the thyroid gland appears to be normal on U/S, then a thyroid function scan is usually not warranted. Moreover, these investigations utilize an intravenously administered radiopharmaceutical compound and should be avoided if possible in the pediatric age group [23]. Thyroid function scans can be reserved for situations where the other diagnostic modalities are inconclusive or if the patient has signs and symptoms of hypothyroidism [16]. In rare cases where a lingual thyroid is suspected, diagnosis can be confirmed by ultrasound or thyroid function scans.
Table 8.3
Differential diagnosis for midline neck mass in children
Thyroglossal duct cyst |
Epidermoid/dermoid/teratoma |
Plunging ranula |
Lymph node |
Thyroid nodule |
Sebaceous cyst |
Thymic cyst |
Vascular malformation |
Third/fourth branchial cleft cyst |
Management
The treatment of a TGDC is complete surgical excision. In his famous manuscript, Sistrunk described the surgical technique for dealing with these lesions. The procedure bears his name and has essentially not changed since 1920. The key to successful removal includes removal of the mid-portion of the hyoid bone in continuity with the cyst and a core of tissue up to the foramen cecum [14]. The recurrence rate using the Sistrunk procedure is estimated to be 4 %. Patients undergoing a cystectomy, without removal of the mid-portion of the hyoid bone, have an average recurrence rate of 50 % with some series reporting rates of 100 % [24]. For the rare occurrence of a TGDC arising in the tongue base, transoral endoscopic removal has been successful [25]. Management of a lingual thyroid is covered elsewhere in this text (Chap. 4).
Lymphangiomas
Embryology
During the third and fourth month of development two paired (jugular and posterior) and two unpaired (mesenteric and cysterna chili) endothelial sacs form the basis of the lymphatic system. These join with lymphatic channels by the end of the ninth week, with cervical lymph nodes forming around the same time [26].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


