CHAPTER 191 Congenital Malformations of the Inner Ear
Development of the inner ear begins early in embryogenesis. By the end of the eighth week, the membranous labyrinth has assumed its characteristic convoluted shape.1 Gradual ossification of the otic capsule develops around the membranous labyrinth and is essentially complete by birth.2 Maturation of the sensory epithelium occurs long after formation of the membranous labyrinth, during the late second and early third trimesters. By weeks 26 to 28 of gestation, hair cell and auditory neural development are largely complete. Thus, the normal human fetus may be able to hear 2.5 to 3 months before birth.
Most inner ear malformations arise when formation of the membranous labyrinth is interrupted during the first trimester of pregnancy.2 This interruption may be either a result of inborn genetic error or a consequence of a teratogenic exposure during the period of inner ear organogenesis between the fourth and eighth weeks of gestation. Genetic errors may be either dominant or recessive and may manifest as sensorineural hearing loss (SNHL) alone or be associated with any of a number of syndromes.4 A partial list of syndromes associated with radiologically detectable inner ear malformation includes Pendred’s, Usher’s, Waardenburg’s, Wildervanck’s (cervico-oculoacoustic), branchio-oto-renal, and Alagille’s sundromes.5–8 Nonsyndromic familial inner ear malformations have also been described.9,10 Teratogenic influences known to affect inner ear organogenesis include in utero viral infection (e.g., rubella, cytomegalovirus infection), chemical teratogens (e.g., thalidomide), and radiation exposure.11 Abnormalities in otic capsule structure and deficiencies in the organ of Corti appear to arise as secondary effects of the earlier error in development of the membranous labyrinth. Derangement of the otic capsule ossification process alone does not appear to be a major mechanism in congenital hearing loss. Ossification of the labyrinthine lumen, however, is a common finding in early acquired deafness, typically arising as a consequence of meningitis.
Developmental damage to cochlear structure and function is not restricted to agents that cause gross structural malformations. Even in doses below those that would be ototoxic in the adult, aminoglycoside antibiotics administered in the animal equivalent of the human first trimester of pregnancy cause severe hearing loss in several species.12 This time frame corresponds to the maturation of the outer hair cells and initiation of the cochlear potentials. Human studies also document this. In 35 of the infants delivered by 72 women who received streptomycin prophylaxis for tuberculosis during the first 4 months of pregnancy, auditory deficits ranging from minor high-frequency threshold elevations to severe bilateral SNHL were noted.13
Incidence
Most children with congenital sensory hearing loss, with the possible exception of auditory neuropathy, would have detectable abnormalities in their inner ears if they could be examined histologically. Because most children with profound bilateral SNHL have radiographically normal inner ears, it can be inferred that malformations limited to the membranous labyrinth predominate. The incidence of malformations reported varies, depending on the hearing level of the study population, the sophistication of imaging used, and the definition of abnormality used by the observer. As a rule of thumb, approximately 20% of cases of congenital SNHL will demonstrate inner ear malformation with modern imaging technology. In a series of 234 children who had SNHL of varying degrees of severity, Reilly14 found cochlear anomalies in only 4% of those evaluated by high-resolution computed tomography (CT). With improvements in CT imaging technology and greater awareness of inner ear deformities, Antonelli and colleagues found anomalies in 31% of 157 children with SNHL of variable degree.15 In another CT study, anomalies were found in 17% of 185 ears of children with SNHL and none in the ears of 309 children without SNHL.16 A large Korean study found 22% incidence of anomalies on radiologic imaging in 590 ears with profound SNHL.17
The incidence of deformities of the semicircular canals (SCCs) and inner ear aqueducts has been less well studied than that of cochlear deformities. In a series of patients with radiographically detectable malformations of the inner ear, the cochlea was involved in 76%, the semicircular canals were involved in 39%, and the vestibular aqueduct (VA) was affected in 32% of ears.3 The total is more than 100%, because many cases demonstrate abnormalities of more than one portion of the inner ear. In recent years, a heightened awareness of VA enlargement, combined with the greater sensitivity of axial CT scans in demonstrating this deformity, has led to a substantial increase in its detection. The rapid accrual of cases by clinicians interested in inner ear malformations suggests that enlargement of the VA will ultimately prove to be the most common radiographically detectable inner ear anomaly.6
Among deaf children with radiographically normal inner ears, histopathologic studies indicate that cochleosaccular dysplasia (Scheibe’s dysplasia) is by far the most common deformity.18 Because of the paucity of pathologic specimens available for examination, it is impossible to estimate the relative frequency of the various membranous malformations.
Classification
The traditional nomenclature used to describe congenital anomalies of the inner ear involves a confusing array of eponyms that stem from the first reports of the various morphologic patterns, usually by 18th or 19th century authors. In this chapter, a descriptive classification system is used, along with the traditional eponymous terminology, to make this topic more logical, easier to learn, and more clinically relevant (Box 191-1). In membranous malformations, this classification is based on histopathologic changes in the inner ear; in combined osseous-membranous deformities, radiographic appearance is used to distinguish among the various entities.3 Correct use of the terminology of pathoembryology is important, although these terms frequently are applied imprecisely in the earlier literature. Key terms are aplasia (complete lack of development), hypoplasia (incomplete development), and dysplasia (aberration in development). The classification scheme used in this chapter has proved to be of practical clinical use and has been employed in most published studies in recent years. Other classification schema that categorize observed anomalies have been proposed.19
Malformations Limited to the Membranous Labyrinth
In malformations limited to the membranous labyrinth, which account for more than 90% of cases of congenital deafness, the bony labyrinth is normal.20–23 In its most severe form, membranous dysplasia involves the entire labyrinth, including the cochlea, semicircular canals (SCCs), utricle, and saccule. A limited form of membranous labyrinthine dysplasia, involving only a portion of the inner ear, also has been described.
Complete Membranous Labyrinthine Dysplasia
Complete membranous labyrinthine dysplasia was first described by Siebenmann and Bing24 and is extremely rare. It has been reported in association with cardioauditory (Jervell and Lange-Nielsen) and Usher syndromes.25
Limited Membranous Labyrinthine Dysplasia
Cochleosaccular Dysplasia (Scheibe’s Dysplasia)
Incomplete development of the pars inferior is the most frequent histopathologic finding in congenital deafness. It was first described by Scheibe26 and commonly is known as cochleosaccular dysplasia. The spectrum of pathologic findings in this anomaly, which is confined to the cochlea and the saccule, has been well described.22,27–30 The organ of Corti is either partially or completely missing. The cochlear duct usually is collapsed, with Reissner’s membrane adherent to the limbus. Less commonly, the duct is distended, presumably as a result of endolymphatic hydrops. The stria vascularis typically is degenerated and may contain colloidal inclusions. Schuknecht31 described characteristic strial changes consisting of aplasia alternating with regions of hyperplasia and gross deformity. Cochlear changes may be severe in the base turn and gradually lessen in intensity toward the apex, or they may be severe throughout. The saccule usually is collapsed and has degenerated sensory epithelium. In cochleosaccular dysplasia, the SCCs and utricle are normal. Auditory neuronal survival is variable but may remain normal into adulthood, at least in some cases. Cochleosaccular dysplasia also has been demonstrated in a number of animal species, including the deaf white cat, Dalmatian dogs, and various mouse mutants.32
Malformations of the Membranous and Osseous Labyrinth
Congenital anomalies of the inner ear that deform the otic capsule are of special interest to the clinician because they may be recognized and differentiated during life through radiographic imaging. As discussed previously, only approximately 20% of congenitally deaf people demonstrate radiographically anomalous inner ears. The clinical manifestations and natural history of these deformities are highly variable. Although some patients are deaf from birth, most maintain some residual hearing into adulthood. Slowly progressive deterioration of hearing during childhood, with eventual stabilization, is common. Sudden decrements in hearing are frequent and may appear to be spontaneous or may be triggered by head trauma, even minor in nature. Presumably, most of these cases are due either to internal fistulization secondary to membrane rupture within the cochlea, with admixture of perilymph and endolymph, or to external fistulization to the middle ear. Fluctuant hearing loss is unusual in these patients, and endolymphatic hydrops is an atypical finding. In some patients, hearing may be best preserved in very high frequencies (greater than 8000 Hz), which are not measured by conventional audiometry.33 Residual ultra-audiometric hearing should be suspected in hearing-impaired children who manifest substantially better auditory function than would be predicted by pure tone results in the speech frequencies.34 Occasionally, malformation of the inner ears may be associated with normal hearing.35,36 This is especially the case for semicircular canal anomalies.37 Vestibular symptoms, which occasionally are severe, are present in approximately 20% of patients.3 Retardation of motor development has been documented in some children with malformed inner ears, particularly those in whom semicircular canals are absent.38 A few patients experience vertigo when exposed to loud sounds—the so-called Tullio phenomenon.39
A wide variety of morphologic patterns of inner ear malformation has been observed radiographically and may involve the cochlea, SCCs, or vestibular aqueduct3,40–45 (Fig. 191-1). Similar diversity has been observed on histologic analysis.21,46–50 A majority of combined osseous and membranous malformations appears to arise from a premature arrest in the development of one or more components of the inner ear (Fig. 191-2). The strongest evidence for this theory comes from the resemblance of most malformed inner ears to the appearance of the inner ear during embryogenesis, particularly between the fourth and eighth weeks of gestation. As a general rule, the earlier the developmental arrest, the more severe the deformity and the worse the hearing will be.
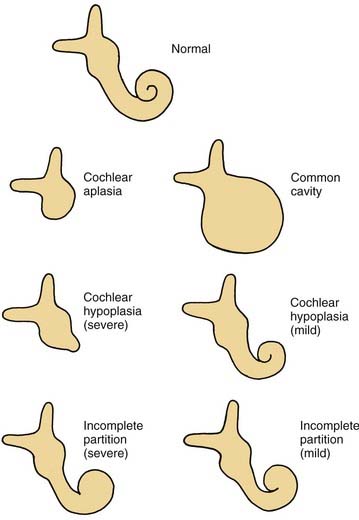
Figure 191-1. Cochlear malformations. Drawings were made from coronal computed tomography scans.
(From Jackler RK, Luxford WM, House WF. Congenital malformations of the inner ear: a classification based on embryogenesis. Laryngoscope. 1987;97[Suppl 40]:2.)
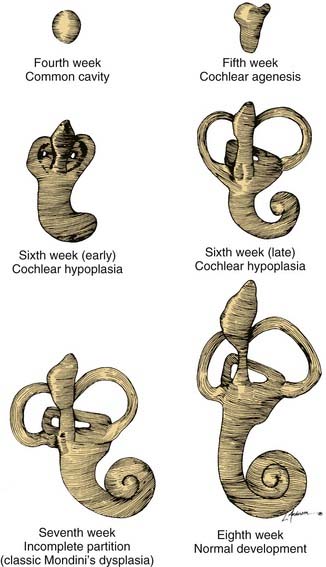
Figure 191-2. Embryogenesis of cochlear malformations.
(From Jackler RK, Luxford WM, House WF. Congenital malformations of the inner ear: a classification based on embryogenesis. Laryngoscope. 1987;97(Suppl 40):2.)
Other anomalies cannot be explained by a premature arrest in development alone and appear to arise from an aberrant embryologic process. An example of this type of anomaly is a cochlea of normal length but abnormal size or coiling geometry.46 In humans, the inner ear is of adult size at birth and shows strikingly little variation in size among individual patients. Pappas and colleagues51 suggested that some children with congenital SNHL and apparently normal inner ear morphology on CT possess subtle abnormalities in the dimensions of inner ear structures. These investigators propose that such dimensional variations arise from a teratogenic insult during the second or third trimester, after the membranous labyrinth has formed but before it has reached adult size. Further study is needed to determine the clinical relevance of these observations.
Whereas some inner ear malformations involve only one portion of the inner ear, many patients have a combination of anomalies involving more than one component. Between the fourth and fifth weeks of development, the spheric otocyst develops three buds that ultimately form the cochlea, SCCs, and VA (see Fig. 191-2). An inner ear malformation may be limited to one of these anlages, may involve a combination of two, or may even affect all three.
The frequent coexistence of deformities involving the cochlea, SCCs, and VA has several possible explanations: (1) the anomaly is genetically predetermined; (2) an insult to the embryo occurred before the fifth week; or (3) each of the buds was susceptible to some teratogenic influence at a later stage of development. A majority of inner ear malformations are bilateral and symmetrical. In cases in which radiographs detect an anomaly on only one side, the opposite “normal” inner ear has a hearing loss in approximately 50% of cases.3
Before the evolution of high-resolution imaging technology, clinicians and histopathologists alike tended to lump these malformations together under the term Mondini’s dysplasia, after the first report by Carlo Mondini. We are indebted to the late Dr. Peter Phelps and Latin scholar Gordon Hartley for an English translation of Mondini’s original article.52 Before the Academy of Sciences of the University of Bologna, Mondini described the inner ear findings in a deaf 8-year-old boy who was struck on the foot by a wagon and later died of gangrene.53 The cochlea possessed only 1.5 turns and had a hollow apical cavity. An enlarged vestibule and VA also were noted. This deformity is the most common form of cochlear anomaly (Table 191-1). Although numerous other distinct anatomic patterns of inner ear malformation are discernible radiographically and histologically, many workers continue to use the term Mondini’s dysplasia to describe them all. To avoid a confusing and overly broad nomenclature system, this designation is best reserved for the particular subtype of cochlear malformation first described by Mondini, whether or not it is associated with other inner ear malformations.
Table 191-1 Relative Incidence of Cochlear Malformations
| Malformation | Incidence (%) |
|---|---|
| Incomplete partition (Mondini’s dysplasia) | 55 |
| Common cavity | 26 |
| Cochlear hypoplasia | 15 |
| Cochlear aplasia | 3 |
| Complete labyrinthine aplasia (Michel’s aplasia) | 1 |
Complete Labyrinthine Aplasia (Michel’s Aplasia)
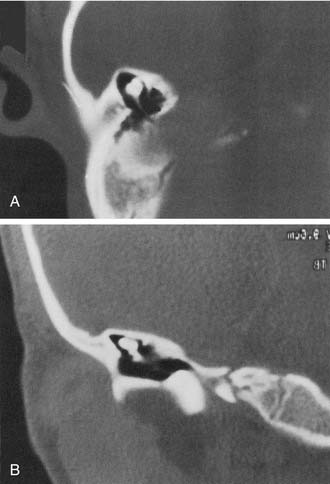
The most severe deformity of the membranous and osseous labyrinth, complete labyrinthine aplasia, was first described by Michel.54 This malformation is exceedingly rare. Presumably, a developmental arrest occurs before the formation of an otic vesicle, resulting in a complete absence of inner ear structures. Complete labyrinthine aplasia has been reported in association with anencephaly and thalidomide exposure.21,55 An association with external ear abnormalities also has been reported.56 A purported case of Michel’s aplasia actually described a cystic inner ear of the common cavity type.57 This is but one example of the inaccurate use of traditional eponyms—a frequent occurrence in the literature. The incidence of complete labyrinthine aplasia is overestimated in the radiographic literature because it is confused with labyrinthine ossification. In the latter condition, which usually is acquired during life, a sizable and dense otic capsule is evident radiographically. In complete labyrinthine aplasia, the otic capsule is entirely absent58 (Figs. 191-3 to 191-5). Such ears are, of course, uniformly deaf.
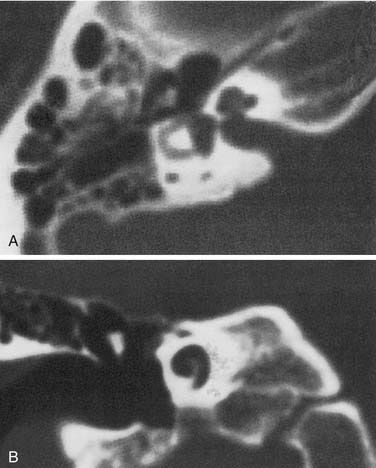
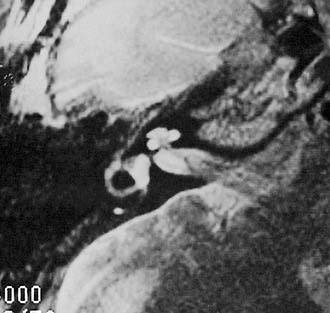
Cochlear Anomalies
Cochlear Aplasia
In cochlear aplasia the cochlea is completely absent, presumably as a result of an arrest in the development of the cochlear bud at the fifth week of gestation (see Fig. 191-1). This morphologic pattern is rare. Radiographically, only a vestibule and SCCs (usually deformed) are present. To differentiate this anomaly from labyrinthine ossification, it is necessary to assess the amount of otic capsule bone anterior to the internal auditory canal (IAC). In cochlear aplasia, the otic capsule is absent, whereas in osseous obliteration, it is dense and of normal dimensions. Ears with cochlear aplasia are devoid of auditory function.
Cochlear Hypoplasia
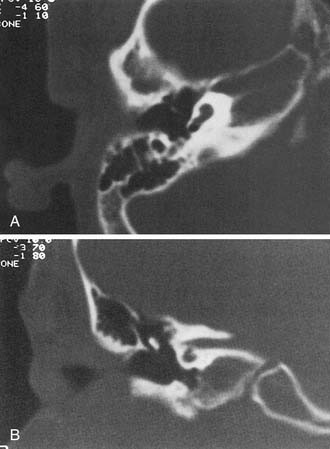
An arrest during the sixth week of gestation results in a hypoplastic cochlea consisting of a single turn or less. This deformity comprises approximately 15% of all cochlear anomalies. Radiographically, a small bud of variable length (usually 1 to 3 mm) protrudes from the vestibule (Fig. 191-6). The vestibule frequently is enlarged, with accompanying semicircular malformations in approximately one half of the cases. Small cochleas lacking a modiolus or other internal architecture have been described histologically.27,50,59 Hearing is variable in these ears and may be remarkably good in view of the minute size of the cochlea. The variability of hearing presumably is accounted for by the degree of membranous labyrinthine development within the truncated cochlear lumen.
Incomplete Partition (Mondini)
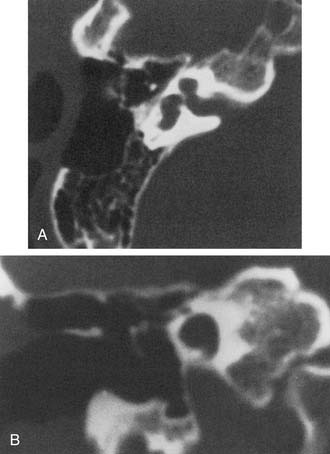
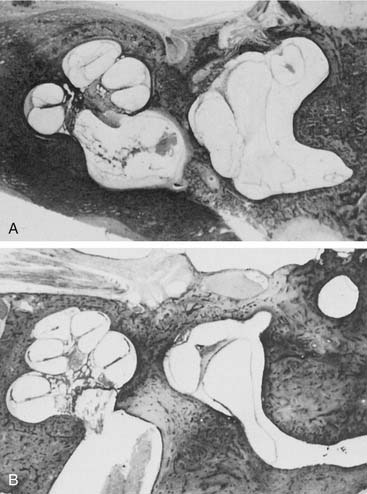
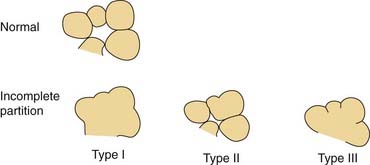
Arrest at the seventh week of gestation yields a cochlea that has only 1.5 turns. This is the most common type of cochlear malformation, accounting for more than 50% of all such deformities. Radiographically, the cochlea is smaller than normal and partially or completely lacks an interscalar septum (Figs. 191-7 and 191-8). Although the cochlea usually measures 8 to 10 mm vertically, it is typically in the 5- to 6-mm range in incomplete partition deformity. Care must be exercised in counting the number of cochlear turns radiographically because this may be difficult to determine even using high-resolution CT. The radiographic diagnosis depends more on cochlear size and the absence of a scalar septum than on the number of cochlear turns perceived. Histologically, incomplete partition appears to be the radiographic correlate of classical Mondini’s dysplasia (Fig. 191-9). In numerous reported cases, a small cochlea with 1.5 turns possessing an apical scala communis secondary to deficiency in the osseous spiral lamina has been described.27,46,48 Sennaroglu and Saatci have subtyped the incomplete partition deformity into three variants60 (Fig. 191-10). Type I lacks the entire modiolus and interscalar septa and demonstrates a cystic appearance. Type II has a normal base turn but a cystic apex (“Mondini type”). Recently Sennaroglu has proposed a type III variant with deficient modiolus and partial interscalar septation at the cochlea’s periphery (L. Sennaroglu, personal communication, 2007). Organ of Corti development is variable, as is the auditory neural population. As might be expected, auditory function also is variable, ranging from normal to profound SNHL. The mean hearing threshold (three-tone average) in a group of 41 ears with incomplete partition was 75 dB.3 SCC deformities accompany incomplete partition of the cochlea in approximately 20% of cases.
Common Cavity
A deformed inner ear in which the cochlea and vestibule are confluent, forming an ovoid cystic space without internal architecture, may be explained by an arrest at the week 4 otocyst stage. Alternatively, this deformity may result from aberrant development at a later stage. An empty ovoid space typically longer in its horizontal dimension is seen radiographically. Although the size of the cyst may vary, it averages 7 mm vertically and 10 mm horizontally. It is quite easy to misdiagnose a dysplastic lateral SCC as a common cavity deformity. The key to differentiating between the two is that a common cavity cochlea lies predominantly anterior to the IAC on axial-plane CT, and a dysplastic vestibular system lies posterior to it. Histologically, an ovoid or spherical smooth-walled cystic cavity containing primordia of the membranous labyrinth has been described.48,50,55 Sensory and supporting cells may be differentiated into recognizable organs of Corti that are scattered peripherally around the walls of the cyst. Neural population usually is sparse or absent. Hearing is usually, but not invariably, poor.
Labyrinthine Anomalies
Semicircular Canal Dysplasia
Dysplasia of the lateral SCC is a common type of inner ear malformation (Fig. 191-11). Approximately 40% of ears with a malformed cochlea will have an accompanying dysplasia of the lateral SCC.3 Occasionally, dysplasia of the lateral SCC exists as the sole inner ear malformation. During the sixth week of development, the budding SCC normally forms a semicircular evagination from the vestibular anlage. The central portion of the pocket-shaped protrusion adheres, leaving a peripheral semicircular tube. When this central adhesion fails to occur, SCC dysplasia results (Fig. 191-12; see also Fig. 191-2). SCC dysplasia occasionally takes the form of a small bud, rather than the more common half-disk shape, presumably because of a slightly earlier timing of the developmental insult. The lateral SCC is deformed more often than the posterior or superior SCC, apparently because it forms earlier in embryogenesis. The typical radiographic appearance of SCC dysplasia is that of a short, broad cystic space confluent with the vestibule (Fig. 191-13).
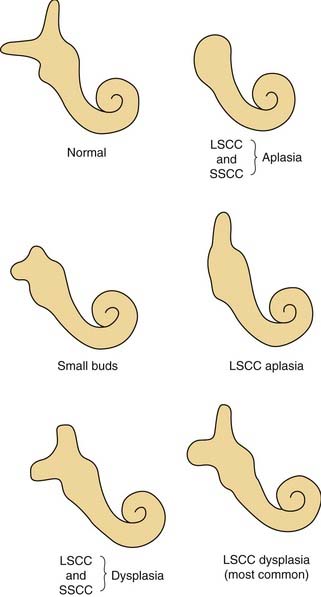
Figure 191-11. Semicircular canal malformations. LSCC, lateral semicircular canal; SSCC, superior semicircular canal.
(From Jackler RK, Luxford WM, House WF. Congenital malformations of the inner ear: a classification based on embryogenesis. Laryngoscope. 1987;97[Suppl 40]:2.)
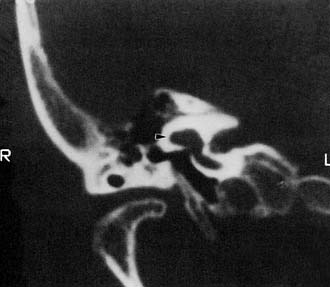
Numerous histologic descriptions of SCC dysplasia exist in the literature.46–50 The half-disk shaped cavity may contain a rudimentary crista ampullaris. The utricle and saccule may be distended, collapsed, or entirely absent. Caloric responses in SCC dysplasia are functionally absent or reduced in most cases, but a few may have normal responsiveness.3 Ears with malformations limited to the vestibular system often have normal or near-normal hearing. When the cochlea also is abnormal, sensory hearing levels tend to be impaired, to a variable degree.62 SCC dysplasia appears to have an association with conductive hearing loss, presumably because of inner ear micromechanical factors rather than stapes fixation.63
Semicircular Canal Aplasia
SCC aplasia is only one-fourth as common as SCC dysplasia.3 It is usually associated with cochlear anomalies.29 Presumably, SCC aplasia arises from a failure in the development of the vestibular anlage before the sixth week of gestation. Most cases are syndromic, with a predominance of the CHARGE association (coloboma, heart defects, atresia of the choanae, retardation of growth and development, genital, and ear abnormalities or hearing loss).64,65
Malformations of the Vestibular and Cochlear Aqueducts
Enlargement of the Vestibular Aqueduct
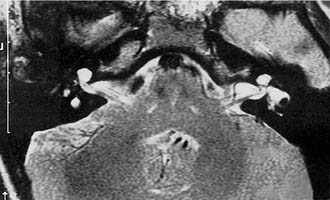
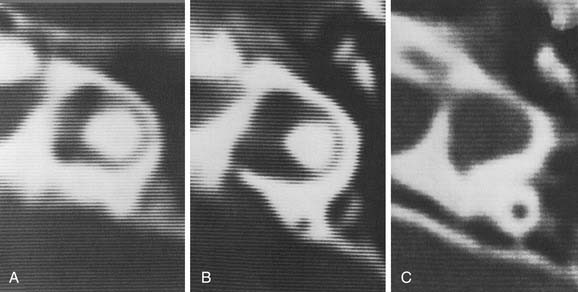
Experience suggests that enlargement of the VA is the most common radiographically detectable malformation of the inner ear.57,66–71 In earlier literature its incidence was underestimated, partly because of a lack of awareness but mostly because enlargement of the VA could be visualized only by lateral polytomography at a time when most studies were confined to the anteroposterior plane. The advent of high-resolution CT in the axial plane has made assessment of the VA much easier. The diameter of a normal VA, when measured halfway between the common crus and its external aperture, is between 0.4 and 1 mm. Enlargement of the VA is diagnosed when its diameter exceeds 2 mm, although enlarged VAs may exceed 6 mm in width. Whereas the VA is well visualized on axial CT (Fig. 191-14), the dilated endolymphatic sac is better seen with T2-weighted magnetic resonance imaging (MRI)72,73 (Fig. 191-15). The sac often is seen to be enormously enlarged on MRI, sometimes measuring 2 cm or more in diameter.74 In many cases, VA enlargement accompanies malformation of the cochlea or SCC. It also may be the sole radiographically detectable abnormality of the inner ear in a child with hearing loss. This condition is commonly referred to as the large VA syndrome, after Valvassori and Clemis’s first descriptions.75,76
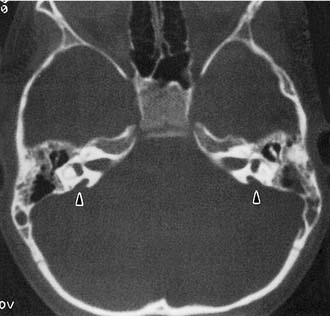
Figure 191-14. Bilateral enlargement of the vestibular aqueducts (arrowheads) as seen on an axial computed tomography scan.
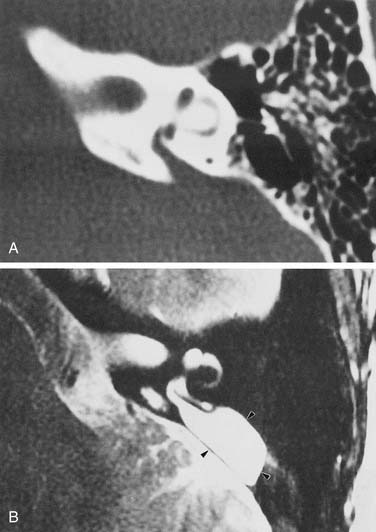
The VA derives from a diverticulum formed in the wall of the otocyst during the fifth week of gestation. It begins as a short, broad pouch but gradually elongates and thins until it achieves its characteristic J shape of adulthood (see Fig. 191-2). A premature arrest in development would be expected to produce a VA that is abnormally short and broad. However, an increasing body of evidence suggests that VA enlargement does not stem from an early arrest of sac development, but rather is an acquired deformity. Histologically, the sac and aqueduct are thin-walled and lack both vascularity and the rugose features thought to be important for physiologic function. Three observations suggest that a large VA stems from an abnormal communication between the subarachnoid space and the fluid chambers of the inner ear. The bone surrounding the VA may show signs of erosion, a finding that is inconsistent with a stable congenital deformity.77 Serial MR images show variability in both the size and signal characteristics of the enlarged sac.78 Presence of cerebrospinal fluid (CSF) under pressure, with consequent “gusher,” within the inner ear has been observed in ears with large VAs during both cochlear implantation and stapedectomy.31,79 Furthermore, the existence of abnormal CSF in the inner ear pathway frequently has been observed with CT or MRI. This anomaly typically consists of a defect of the cochlear modiolus at the distal end of the IAC.12,73
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


