Chapter 136 Congenital Hypertrophy of the Retinal Pigment Epithelium
Introduction
Congenital hypertrophy of the retinal pigment epithelium (CHRPE) is generally an asymptomatic congenital hamartoma that occurs in three variant forms: either as solitary, or grouped or multiple pigmented fundus lesions. Lesions are usually observed during routine ophthalmoscopy.1 Multiple CHRPE may be associated with familial adenomatous polyposis (FAP), an autosomal dominant disease with numerous adenomatous polyps of the colon and rectum. FAP with prominent extracolonic manifestations has been termed Gardner syndrome (GS) or another variant as Turcot syndrome for FAP with brain tumors lesions (Fig. 136.1). The observation of CHRPE in a premature child provides evidence for the congenital nature of this lesion.2
Epidemiology/demographics
The reported prevalence of CHRPE is as low as 3 described cases among 2400 ophthalmic examinations (1.25%). The prevalence of FAP varies from 1 in 7000 to 1 in 22 000 individuals. Approximately 70–90% of FAP patients have pigmented fundus lesions.3
Clinical findings and classification
Solitary CHRPE
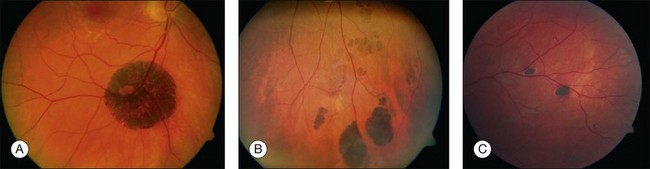
Solitary CHRPE is typically a flat, round, hyperpigmented lesion with smooth or scalloped margins that is well-demarcated from normal-appearing retinal pigment epithelium (RPE) (Fig. 136.2A). The color may vary from light gray, brown to black and is independent of the patient’s race.1 A marginal halo of depigmentation may surround the lesion and “punched-out” inner lacunae with hypopigmentation may be present in larger lesions. Both depigmented zones show the tendency to progress gradually over time, and eventually, may involve the entire lesion.4–7 The overlying retina and its vasculature appear normal in most cases, however discrete focal intraretinal pigmentation may become visible near the margin on biomicroscopy.4 Some lesions may be associated with retinal vascular abnormalities including capillary and large-vessel obliteration, microaneurysmal changes, chorioretinal anastomoses, and neovascularization. The size may vary from 100 µm to several disc diameters, occasionally occupying an entire quadrant. Lesions may be found anywhere in the fundus, with predominance in the superotemporal and equatorial region while the macula is rarely involved. Solitary CHRPE enlarge in 46–83% of cases over three or more years of follow-up. Extension into the fovea may result in reduced visual acuity.8 Some heavily pigmented nodular lesions may develop within CHRPE.9 Histopathologically nodular lesions proved to be adenocarcinomas in two reports.10,11 Untreated nodular lesions may enlarge to form pedunculated tumors of >7 mm thickness with serous retinal detachment.11
Grouped CHRPE
When several lesions of varying size are arranged in a cluster, resembling the footprint of an animal (‘bear tracks’), they are named “grouped CHRPE” (233800, OMIM). Grouped lesions are flat, well-demarcated round oval or geographic black spots with increasing size towards the fundus periphery. Each cluster includes approximately 3–30 lesions, which may vary in size from 100 to 300 µm lesions (Fig. 136.2B). Etiology and development of grouped CHRPE remain unknown. The presence of grouped CHRPE and additional ipsilateral sectorial pigmented skin lesions following the “cutaneous lines of Blaschko” gave evidence for possible pigmentary mosaicism in both the eye and skin.12 Therefore, grouped CHRPE are considered as a cluster of atypical hyperpigmented RPE cells, that derive from the edge of the optic nerve and migrate along the stream of embryologic tissue lines, analogous to the “cutaneous lines of Blaschko.” The sectorial pattern of grouped CHRPE obviously reflects the stream, outgrowth and migration path of RPE cells during embryogenesis.13 Bilateral grouped CHRPE has rarely been observed. There are no systemic associations in patients with solitary and grouped CHRPE.
Multiple CHRPE
Multiple CHRPE lesions in FAP are generally smaller (50–100 µm in diameter) compared with solitary CHRPE. They are black, brown, or light gray (Fig. 136.2C). Larger lesions may be surrounded by a depigmented halo, mottled RPE,14 window-defect-type changes adjacent to retinal vessels,15 may contain depigmented lacunae, and can be accompanied by small pigmented satellite lesions. Retinal invasion and glial, capillary and pigment epithelial proliferation and hypertrophy are typical. More than four widely spaced lesions per eye or bilateral involvement are suggestive of FAP.16 Traboulsi et al. examined 16 GS families for multiple CHRPE. Of 41 GS patients, 37 (90%) had multiple CHRPE lesions.14 Lesions were bilateral in 32 patients (78%). The presence of bilateral lesions or multiple unilateral lesions (>4) appeared to be a useful clinical marker (specificity, 0.95; sensitivity, 0.78) for GS. In view of the multiple, bilateral character of retinal lesions observed in FAP, they can be considered a clinical disease marker. However, the absence of CHRPE has no predictive value for absence of GS or FAP.
Differential diagnosis
Often CHRPE has been misdiagnosed as choroidal malignant melanoma. The latter is almost always elevated, less homogeneously pigmented and less sharply demarcated compared with CHRPE, and usually exhibits growth in all three dimensions. Choroidal nevi are flat and located below the RPE. Their color may vary from light to dark brown. The borders are less well demarcated and often are feathery because nevus cells extend along larger choroidal vessels. Frequently, drusen and pigmentary mottling are present on the surface of nevi. Melanocytomas of the choroid have a similar appearance to CHRPE, except for their homogeneously black color. Black sunburst lesions in sickle cell retinopathy may appear as dark gray to brown convex-shaped lesions predominantly in the midperiphery. True hyperplasia of the RPE has ill-defined borders and invades the retina, often leading to retinal distortion. Focal pigmentation caused by injury, inflammation, or drug toxicity may resemble CHRPE but can be differentiated on the basis of a more irregular shape, widespread distribution, and associated clues suggesting acquired disease.17
Associated extraocular findings
Solitary, unilateral lesions and grouped CHRPE are restricted to the RPE with no other ocular or extracellular findings.18 Multiple or bilateral CHRPE may be associated with autosomal dominant FAP or GS (175100, OMIM)). Gardner syndrome is characterized by FAP of the large and small intestine, hamartomas of the skeleton and various soft-tissue tumefactions.19,20 Intestinal polyps generally appear during the third decade of life and invariably progress to adenocarcinomas by the fifth decade. Skeletal hamartomas, most commonly benign osteomas of mandible skull, orbits and long bones, become apparent in the third decade of life, as well as soft-tissue abnormalities including epidermoid and sebaceous cysts, dermal fibromas, lipomas, leiomyomas, desmoid, and mesenteric fibromatosis. Further extracolonic manifestations of GS include tumors of the thyroid, adrenal, and bladder, as well as sarcomas and hepatoblastomas. Turcot syndrome (233800, OMIM) is associated with FAP and brain tumors, e.g., astrocytomas, medulloblastomas, and ependymomas.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


