CHAPTER 202 Congenital Disorders of the Larynx
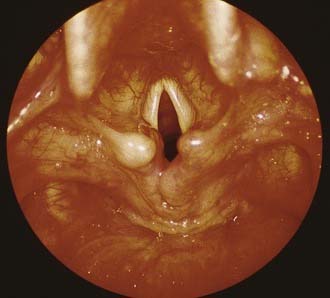
Dynamic evaluation of the larynx by flexible laryngoscopy is best performed in the patient who is awake. Although at least a 2.5-mm diameter fiberoptic laryngoscope is recommended for optimal viewing, fiberoptic laryngoscopes with diameters as small as 1.9 mm allow the neonatal larynx to be viewed with a minimum of trauma to the young patient. Direct laryngoscopy of the neonatal larynx with a telescope or microscope provides the best optics (Fig. 202-1). To fully evaluate the larynx, the endoscopist should note findings in the vallecula, piriform fossae, postcricoid region, arytenoids, interarytenoid area, aryepiglottic folds, epiglottis, false vocal cords, ventricles, true vocal cords, and the subglottis.
Laryngomalacia
Without question, the most common cause of stridor in infants is laryngomalacia, a disease more commonly found in term infants, and in males more than females.1 The newborn with laryngomalacia typically develops intermittent, inspiratory stridor within the first 2 weeks of life, which resolves slowly over several months. Many authors describe the stridor as high-pitched, but compared to the stridor of vocal cord paralysis it is relatively low-pitched and does not have a musical quality. The stridor nearly always worsens with feeding, and often the baby will need to take breaks while feeding in order to breathe. The stridor of mild laryngomalacia often improves with crying as tone in the pharynx is increased. Conversely, in moderate-to-severe laryngomalacia, the stridor typically will worsen with crying because of the increased airflow through the severely collapsed larynx. Laryngomalacia may be an isolated finding in the otherwise healthy infant or may be associated with other neurologic disorders such as cerebral palsy.
The median time to spontaneous resolution of stridor is 9 months of age, and 75% will have no stridor by 18 months of age.2 Infrequently, the laryngomalacia is severe, resulting in feeding difficulties, failure to thrive, apnea, pectus excavatum, or cyanosis. In these cases, surgical intervention is recommended to prevent worsening failure to thrive, cor pulmonale, and cardiac failure.1–3
The inspiratory stridor of laryngomalacia results from collapse of the supraglotttic larynx, creating a narrow airway and turbulent airflow (Fig. 202-2). The etiology of this collapse has been elusive but it appears to be related to neuromuscular hypotonia. Sensorimotor integration of peripheral sensory afferent reflexes, brainstem function, and the motor efferent response are responsible for laryngeal function and tone.1 The laryngeal adductor reflex (LAR) is a vagal nerve-mediated reflex activated by sensory stimulation of the mechanoreceptors and chemoreceptors of the superior laryngeal nerve located in the region of the aryepiglottic fold. Dysfunction anywhere along the afferent, brainstem, or efferent pathway of the LAR can result in altered laryngeal tone and function. Infants with laryngomalacia have been found to have elevated laryngopharyngeal sensory testing thresholds, showing that the sensorimotor integrative function of the larynx is altered, likely leading to the weak laryngeal tone seen in infants with laryngomalacia.1
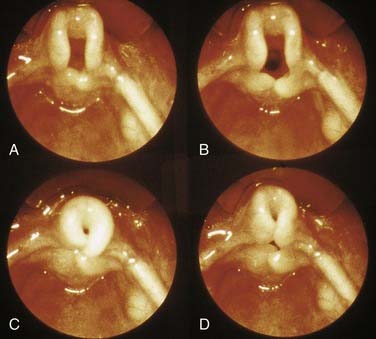
Figure 202-2. Laryngomalacia. Progressive airway obstruction on inspiration. Note omega-shaped epiglottis.
(From Benjamin B. The pediatric airway, slide #2, Slide Lecture Series. American Academy of Otolaryngology–Head and Neck Surgery, 1992.)
In several series, a high prevalence of gastroesophageal reflux disease (GERD) has been reported in patients with laryngomalacia (50% to 100%).1,4–6 When present, GERD can cause airway edema and thus contribute to the airway compromise. Children with laryngomalacia should be evaluated for GERD and if airway surgery is needed, care should be taken to control GERD before surgery to avoid GERD-induced delayed healing.6 Mild GERD may improve with the treatment of laryngomalacia when the negative intrathoracic intraesophageal pressures decrease. Conversely, treatment of GERD may improve laryngomalacia as acid ceases to irritate the larynx and edema resolves.5 In the child with GERD related to airway obstruction, the GERD can be expected to improve significantly after aryepiglottoplasty.7
The diagnosis of laryngomalacia requires an endoscopic examination; however, the optimal type of endoscopic examination is somewhat controversial. If the patient is sedated and undergoes fiberoptic flexible laryngoscopy (FFL), the topical lidocaine typically used can cause increased collapse of the arytenoids and folding of the epiglottis during inspiration—possibly leading to overestimation of the severity of laryngomalacia.8 These findings suggest that the optimal way to diagnose laryngomalacia is with FFL in the awake patient. Conversely, the awake technique has been found to miss mild laryngomalacia or lead to overdiagnosis in the patient with a normal airway.9 The advantages of awake FFL compared to FFL under general anesthesia include the ability to perform the examination expediently in the clinic setting with the parents present, without the need for sedation, leading to less medical expense. The primary disadvantage of the awake FFL is that in nearly all cases the infant is crying during the examination, which can alter the appearance of the supraglottic structures. An omega-shaped epiglottis is often associated with laryngomalacia but can also be found in otherwise normal infants with no airway obstruction. If fluoroscopy is performed, downward anterior displacement and bowing of the aryepiglottic folds, horizontal infolding of the epiglottis, and in some cases, narrowing of the trachea are seen on inspiration, all of which reverse on expiration.10
Approximately 17% of infants with laryngomalacia have a second synchronous lesion that may lead to airway difficulties.11 The decision to perform a bronchoscopy in patients who present with typical laryngomalacia is also controversial. Some contend that bronchoscopy need be done only in select infants with laryngomalacia who present with “apnea, failure to thrive, or features inconsistent with isolated laryngomalacia.”2,12 Others strongly recommend a complete evaluation of the tracheobronchial tree in all symptomatic infants to avoid missing life-threatening lesions.13
The anatomic abnormality causing the supraglotttic obstruction of laryngomalacia varies among infants. Most have anterior prolapse of the mucosa overlying the arytenoid cartilages (57%); others have short aryepiglottic folds that tether the epiglottis posteriorly (15%), posterior collapse of the epiglottis (12%), or some combination of these findings (15%).2 Several classification systems have been proposed with none being predominant at this time.2,14,15
Approximately 10% to 31% of infants seen by a pediatric otolaryngologist require surgical intervention for their laryngomalacia.1 Surgery is indicated if there is feeding difficulty with failure to thrive, cyanosis, apnea, respiratory distress, or hypoxia. The current standard treatment is supraglottoplasty (aryepiglottoplasty) (Fig. 202-3). The surgical resection can be performed using the carbon dioxide (CO2) laser, laryngeal microscissors, or microdébrider.1–3,5,16 Various types of supraglottoplasty have been described based on what portion of the prolapsing supraglottis is removed: the tissue overlying the arytenoids (with care taken to preserve the interarytenoid mucosa), the aryepiglottic folds, or the posterior portion of the epiglottis.5 Unilateral supraglottoplasty has been advocated by some authors to relieve the airway obstruction while reducing the risk of postoperative supraglottic stenosis.17,18
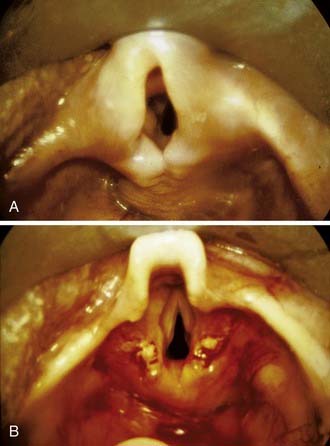
Short aryepiglottic folds that tether the supraglotttic structures in close anterior and posterior approximation can be divided or excised.19 If the epiglottis is displaced posteriorly, an epiglottopexy can be performed.20 In this procedure the mucosa of the lingual surface of the epiglottis and the corresponding mucosa of the base of the tongue are removed with the CO2 laser, then the epiglottis is sutured to the tongue base. These procedures are well tolerated by the infants, typically requiring only a short 1- to 3-day hospital stay. The success of the various forms of supraglottoplasty-epiglottopexy are high, with the vast majority of children obtaining relief from their obstruction.3,16,19–23 Those children who do not improve with supraglottoplasty usually have underlying neurologic or syndromal abnormalities or associated tracheomalacia and may require a tracheotomy.22,23 Complications are rare, with the most feared being supraglottic stenosis, which may occur in up to 4% of cases.22 More frequently, transient dysphagia and aspiration can occur in approximately 10% to 15% of patients.1
Although laryngomalacia is typically thought of as occurring only in infants, it is occasionally observed in older children and adults. Neurologically impaired children (i.e., those with cerebral palsy) with poor pharyngeal control can develop laryngomalacia. Exercise-induced laryngomalacia results when enough inspiratory force occurs during exercise to draw the aryepiglottic folds into the larynx, partially obstructing the glottis.24,25 In severe cases with redundant aryepiglottic folds, supraglottoplasty can be beneficial. Similarly, if the epiglottis is elongated and flaccid, a partial epiglottidectomy can be effective.26
Vocal Fold Paralysis
Vocal fold paralysis (VFP) is another common cause of neonatal stridor. The stridor is inspiratory or biphasic, with a high-pitched musical quality. VFP in a newborn may be idiopathic or can result from birth trauma, central or peripheral neurologic diseases, or thoracic diseases or procedures.27,28 The diagnosis is best made by FFL in the awake patient so that the effect of anesthesia on vocal fold function is minimized.
Iatrogenic left VFP may be present following thoracic surgery, particularly patent ductus arteriosus (PDA) ligation or repair of an interrupted aortic arch. In these infants, the left recurrent laryngeal nerve is damaged at the point that it passes around the ductus arteriosus (unless a right aortic arch is present). The incidence of recurrent laryngeal nerve injury is estimated at 1% to 7.4% of PDA ligation cases.29,30 Most infants with a unilateral VFP are brought to the attention of the otolaryngologist for evaluation of a weak cry or stridor or for inability to wean the infant off ventilatory support if thoracic surgery was performed.30,31 Of those children evaluated by a pediatric otolaryngologist with a unilateral vocal fold paralysis after cardiac surgery approximately 35% will recover vocal fold function.31 At least half of these patients will have swallowing dysfunction, usually aspiration or laryngeal penetration with modified barium swallow.31,32 Although many of the patients with a unilateral vocal fold paralysis will be able to feed orally, many will initially require a gastrostomy tube.31,32
The more difficult problem is bilateral VFP. The degree of airway obstruction is often severe, requiring a tracheotomy in up to 73% of children.33–35 The diagnosis of vocal fold paralysis can be made on FFL, but differentiation between bilateral vocal fold fixation and neurogenic VFP cannot be made unless direct laryngoscopy is performed and the glottis palpated. Laryngeal electromyography in the pediatric patient under a light plane of anesthesia can also be helpful to differentiate fixation vs paralysis and assist with prognosis.36,37
Workup of patients with idiopathic VFP should include a magnetic resonance imaging (MRI) scan of the brain to rule out an Arnold-Chiari malformation or other intracranial cause of brainstem compression.38–40 If such a malformation is identified, then early decompression is recommended to obtain a better outcome.40 If idiopathic congenital bilateral VFP is present, a genetics consultation is recommended to evaluate the patient for chromosome abnormalities.41
Approximately 70% of noniatrogenic unilateral vocal fold paralyses will resolve spontaneously, most within the first 6 months of life.27 The patient with unilateral VFP is often able to compensate for purposes of feeding without medical intervention. Sometimes an infant will require nasogastric feeds on a short-term basis or a gastrostomy tube. An occupational or speech pathology feeding consult is helpful with these infants to rule out aspiration (as seen on modified barium swallow) and to counsel the parents on how to feed the patient. Most often, these infants should start with a slow-flow nipple, then work up to a regular nipple. The voice of most infants with a unilateral VFP is quiet but audible and gains strength with time.
Occasionally, the child with unilateral VFP has problems with severe aspiration and dysphonia. In these children, injection of the vocal fold can be helpful. Most commonly, injection materials used include cadaveric dermis (Cymetra), calcium hydroxyapatite (Radiesse or Radiesse Voice Gel), hydrated porcine gelatin powder (Surgifoam), or absorbable gelatin sponge (Gelfoam), or autologous fat.42,43 Alternatively, in the older child a medialization laryngoplasty (thyroplasty) can be performed.42–45 The retrospective review by Link and others of 12 type I thyroplasty procedures in eight pediatric patients ages 2 to 17 years demonstrated that the anatomically lower position of the vocal fold (compared with that in adults) must be taken into consideration if optimum outcomes are to be obtained in children.45
The vocal folds spontaneously become mobile in up to 65% of patients with bilateral VFP.27,46 Prognosis is better if there are no associated anomalies.33 Most recovery of vocal fold function occurs within 24 to 36 months, although it has been reported in patients up to 11 years of age.33,34 If recovery occurs after 2 to 3 years, it is often incomplete because of laryngeal muscle atrophy, cricoarytenoid fixation, and synkinesis.33 The timing of possible surgical intervention is debated. Some authors recommend intervention at a young age. Others suggest waiting until adolescence when the patient is able to decide.33,34,47
Many surgical options are possible to improve the airway of a patient with bilateral vocal cord paralysis.48 The plethora of surgical options indicates that none is uniformly successful—particularly in the young patient. Endoscopically, a lateral cordotomy can be performed to increase the glottic area.49,50 An arytenoidectomy can be performed via an endoscopic or external cervical approach or using the Woodman approach.33,51 Alternatively, an arytenoidopexy can be performed using a laterocervical approach.27,52,47 Triglia and colleagues performed an arytenoidopexy on 34 children with bilateral vocal cord paralysis who had a mean age of 20 months.47 Of the 15 children who had a tracheotomy or endotracheal tube (ETT) before the procedure, 14 were successfully decannulated postoperatively. Another option is to expand the cricoid cartilage by placing a costal cartilage graft posteriorly, thereby separating the arytenoids and increasing the area between the vocal folds. This procedure can be performed with a laryngofissure or endoscopic approach.53,54 Finally, lateralization of the paralyzed cord has been described as a potentially reversible approach to vocal cord paralysis.55
Hartnick and colleagues described 52 children who underwent surgery for bilateral VFP.56 The procedures most successful in realizing tracheotomy decannulation were those that included vocal cord lateralization procedures combined with a partial arytenoidectomy (71% decannulation rate). These procedures were more successful than CO2 laser cordotomy or arytenoidectomy procedures (29% decannulation), for isolated arytenoidopexy procedures (25% decannulation) or for posterior costal cartilage graft procedures (60% decannulation). No prospective randomized studies have been performed to evaluate the outcomes of these different surgical options, although meta-analysis has suggested that external arytenoidopexy and external arytenoidectomy are more effective than carbon dioxide ablation procedures.57
Laryngeal Web/Atresia
Posterior Webs
Apparent bilateral vocal cord paralysis may occur if an interarytenoid web is present in the posterior larynx causing limited vocal fold abduction (Fig. 202-4). This type of congenital web is rare and often necessitates a tracheotomy in the early years of life.58 Stridor is the major presenting clinical feature, but patients can also present with obstructive cyanosis at birth or with episodes of apnea.
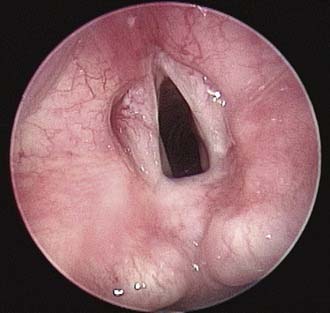
Diagnosis is made at the time of microscopic laryngoscopy with palpation of the interarytenoid area. Posterior webs may be associated with subglottic stenosis, and bulky arytenoids and abnormal laryngeal position are often present. Management options include long-term tracheotomy with probable decannulation after 3 to 5 years, division of the web (frequently unsuccessful), arytenoidectomy, or open airway reconstruction with a posterior cartilage graft.58
Anterior Webs
Anterior congenital laryngeal webs are also rare anomalies typically diagnosed in the neonatal period after an investigation for the source of aphonia and stridor (Fig. 202-5). Occasionally a minor web will not be diagnosed until the child is older and undergoes an evaluation for chronic hoarseness. Most commonly, a web will cause partial obstruction of the anterior glottis and will extend into the subglottic area, causing congenital subglottic stenosis.59
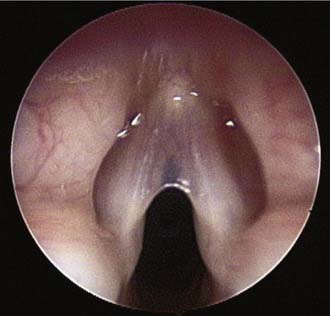
Figure 202-5. Anterior glottic web. Patient presented with chronic hoarseness and mild stridor.
(Copyright Peter Koltai, MD.)
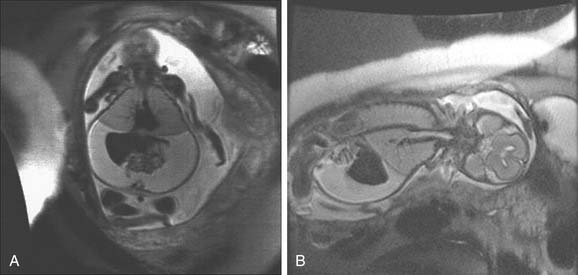
The most severe form of a laryngeal web is total atresia of the larynx (Fig. 202-6). A fetus with laryngeal atresia or a web obstructing nearly all of the larynx will develop congenital high airway obstruction syndrome (CHAOS). Magnetic resonance imaging of the affected fetus will show enlarged echogenic lungs, inverted diaphragms, massive ascites, and dilated airways (Fig. 202-7).60,61 Death results in these infants unless a tracheotomy is performed at the time of birth, therefore delivery by ex utero intrapartum treatment (EXIT procedure) is recommended. The EXIT procedure establishes an airway before completion of birth, while the neonate remains on placental support.
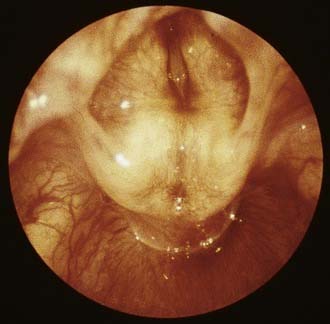
Figure 202-6. Laryngeal atresia.
(From Benjamin B. The pediatric airway, slide #33, Slide Lecture Series. American Academy of Otolaryngology–Head and Neck Surgery, 1992.)
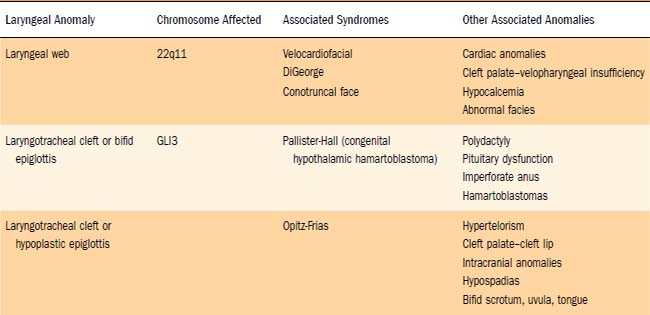
Many anterior laryngeal webs are associated with deletions of chromosome 22q11.63 In a review of 17 patients with anterior glottic webs, 11 (65%) were found to be positive for chromosome 22q11.2 deletion.63 These same microscopic and submicroscopic deletions cause a wide range of phenotypes including DiGeorge’s syndrome, velocardiofacial (Shprintzen) syndrome, conotruncal anomaly face syndrome, and sporadic or familial heart defects (Table 202-1). The acronym CATCH 22 (cardiac defect, abnormal facies, thymus hypoplasia, cleft palate, hypocalcemia, and chromosome 22 deletion) has been used to describe the various phenotypes. For this reason, all patients with a diagnosis of laryngeal web should undergo genetic screening and a thorough cardiovascular evaluation, with particular attention being paid to the aortic arch. Delayed diagnosis of a vascular ring can occur in the patient with a laryngeal web whose stridor and dysphagia are attributed only to the presence of the web.62
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


