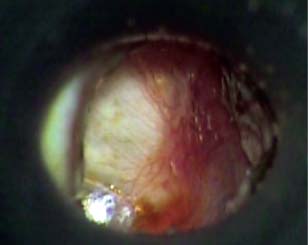
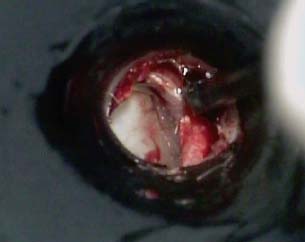
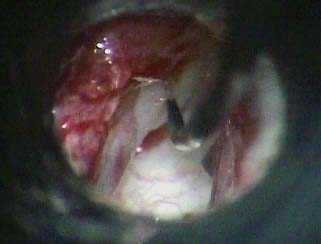
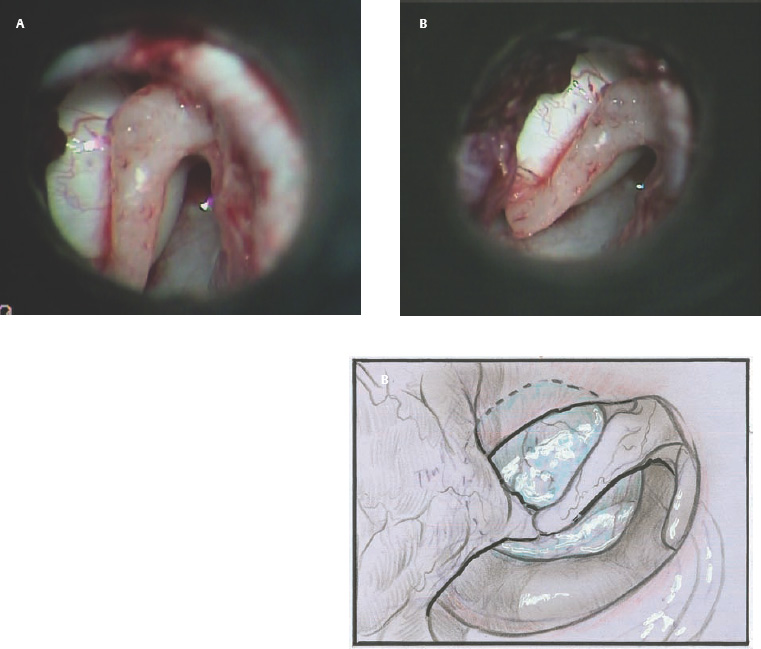
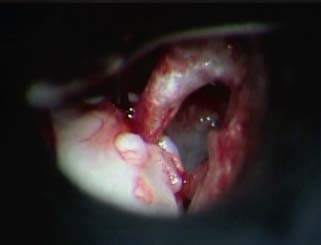
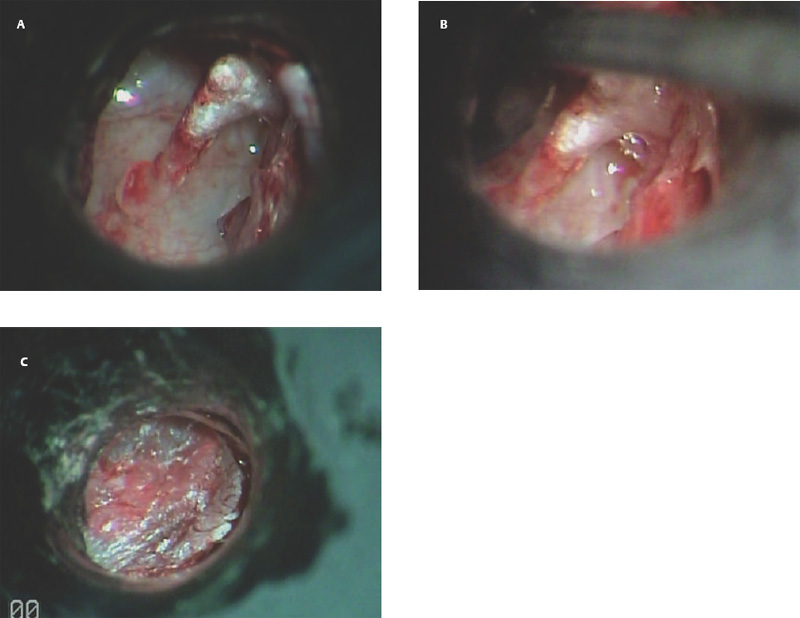
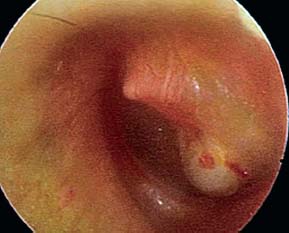
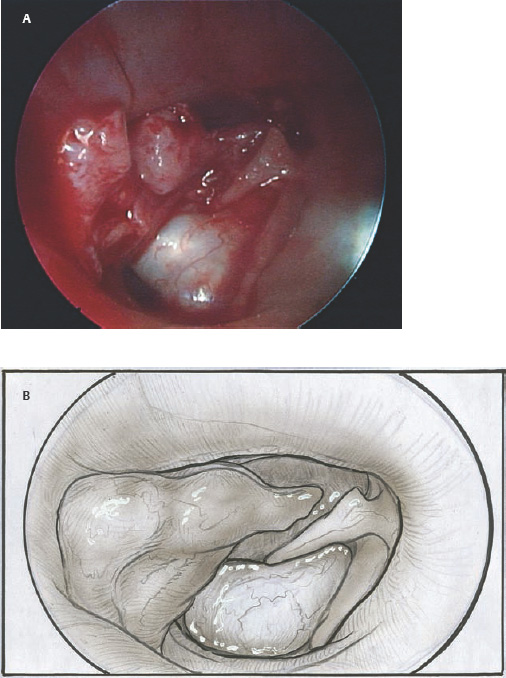
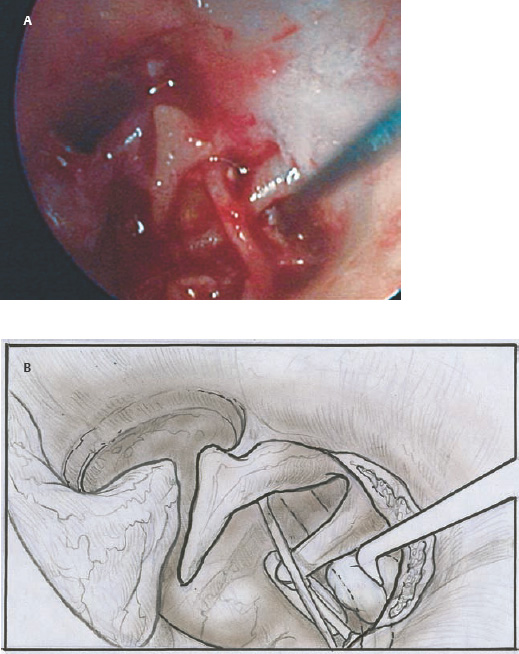
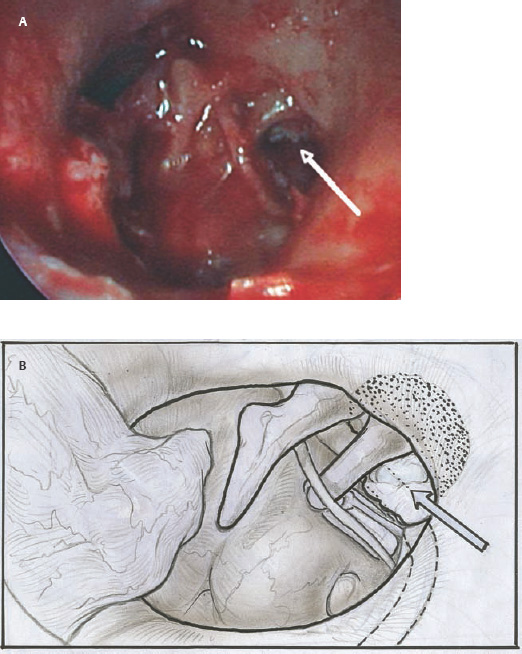
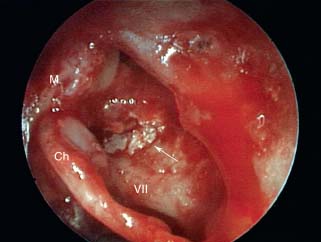
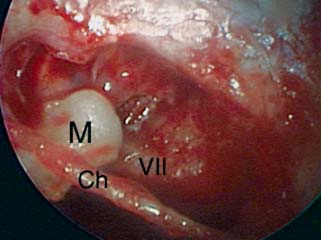
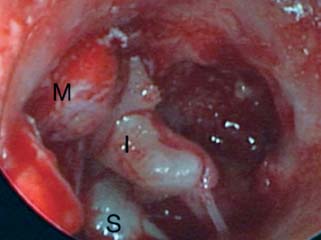
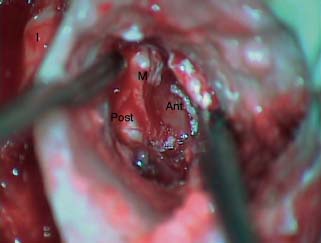
9 Congenital Cholesteatoma Congenital cholesteatoma is cholesteatoma that occurs in a child, behind an intact tympanic membrane.1 Congenital cholesteatoma is thought to arise from an epithelial rest that gets trapped in the middle ear cleft in utero, and that enlarges over time to form a ball of keratinizing squamous epithelium.2 This entity is truly, then, an epidermoid tumor, rather than the end-product of an inflammatory process, although Tos has argued the contrary.3 Congenital cholesteatomas are usually discovered in infants or young children on routine physical exam. They appear as a white mass behind an intact tympanic membrane (Fig. 9.1). The average age at presentation is ∼4 years. In the original description of congenital cholesteatoma by Derlacki and Clemis,4 the diagnosis required that the ear be otherwise completely normal, with no history of otitis media. Because the incidence of otitis media is high in the general population of children, however, a history of ear infection or the presence of a middle ear effusion should not disqualify the diagnosis, as long as there is no history of otorrhea and the tympanic membrane is free of perforation or retraction. In fact, a certain number of congenital cholesteatomas will first be discovered during a myringotomy for serous otitis. Congenital cholesteatomas begin in the middle ear in two general locations—in the anterosuperior quadrant (Fig. 9.1) or in the posterosuperior quadrant. The former are usually associated with an intact ossicular chain and, if detected early enough, can be removed completely with the expectation of a normal hearing outcome. The latter often involve the incus and stapes early and are less likely to result in normal hearing. Congenital cholesteatomas that are missed in early childhood and that are allowed to become sizable will behave no differently from acquired cholesteatomas—they will erode bone as they grow, and they will eventually perforate and drain.5 Congenital cholesteatomas are almost always unilateral; however, bilateral congenital cholesteatomas have been described.6 The treatment of congenital cholesteatoma is surgical. These should be operated on when first discovered, to prevent the potential problems associated with continued growth. Anteriorly situated cholesteatomas can usually be removed via an extended tympanotomy approach, as described by Levenson et al.1 A transmeatal approach can usually be employed, even in younger children, but it is acceptable to use a postauricular incision to gain adequate exposure if the meatus is very small. A standard tympanomeatal incision is made in the posterior canal skin, and is extended anteriorly to ∼3 o’clock in the right ear, or 9 o’clock in the left ear, to create a flap that is pedicled to the anterior-inferior canal skin (Fig. 9.2). After elevating the flap, the drum is separated from the manubrium of the malleus (Fig. 9.3). This is the key step in gaining anterior exposure. This is done by incising the investing mucosa along the length of the malleus with a sharp pick and peeling the drum epithelium downward, like a stocking, toward the umbo. The fibrous insertion of the drum to the umbo can be incised with a Bellucci scissors—this allows the flap to be reflected forward and downward to gain maximal exposure of the anterior middle ear space. Fig. 9.2 Tympanomeatal incision is extended anteriorly to 9 o’clock (for left ear), creating a flap that will be pedicled anteroinferiorly. This approach should be sufficient to expose the anterior and inferior margins of the cholesteatoma sac (Fig. 9.4A,B). The superior margin may extend upward to the attic and can be exposed by removing the edge of the scutum with a stapes curette. The cholesteatoma is enveloped by a very thin layer of normal mucosa. This mucosal layer is incised along the inferior margin of the sac, and the lesion is freed by blunt dissection using a sweeping motion with a whirlybird (Hough excavator) along the anterior and superior borders, keeping the capsule intact (Fig. 9.5). The lesion is usually attached to the tensor tympani tendon near the processus, and should be separated sharply from the tensor using a Rosen needle. The cholesteatoma can usually be delivered in toto once this is done. Fig. 9.5 The mucosal envelope is incised, and the lesion is completely mobilized. Occasionally the lesion cannot be removed without rupturing the sac. If this does occur, the contents (debris) should be carefully evacuated with suction and cup forceps, care being taken not to spill the contents of the lesion into the middle ear, and the capsule should be maintained as completely as possible on the medial (deep) surface. Once the lesion is reduced in size, the capsule can be dissected and the remainder of the sac delivered. Once the lesion is removed, it is important to inspect the attic and protympanum using a mirror or endoscope to be sure that there is no residual disease (Fig. 9.6A–C). It is also advisable to scrape the anterior surface of the tensor tendon with a sharp pick to be certain that no epithelium is left there. The tympanomeatal flap can be replaced and the external canal packed lightly with Gelfoam (Pfizer, Inc., New York, NY). The ear should heal normally, with normal hearing resulting. Fig. 9.7 Endoscopic photograph of posteriorly situated congenital cholesteatoma of the left ear. Posteriorly situated cholesteatomas (Fig. 9.7) cannot usually be removed in toto because they usually involve the incus. In this respect they must be handled more like acquired cholesteatomas. After an extended tympanomeatal flap is raised (Fig. 9.8A,B), the posterior and superior bony overhang is removed with a curette, and the extent of the lesion is evaluated (Fig. 9.9A,B). The incus must usually be separated from the stapes and removed if it is enveloped by the disease (Fig. 9.10A,B). If there is considerable superior extension to the epitympanum (Fig. 9.11), a transcanal atticotomy can be performed, removing the scutum with a curette until the posterosuperior margin is identified. The sac is dissected from the medial attic wall toward the middle ear and delivered (Figs. 9.11 and 9.12). Primary ossicular reconstruction can be undertaken, as shown here, or delayed until a second stage (Fig. 9.13). Large congenital cholesteatomas are handled in the same way as large acquired cholesteatomas. A canal wall up mastoidectomy is created to assess the posterior extent of the disease. A transcanal atticotomy is performed to remove the disease from the epitympanum, and a facial recess opening to dissect the sac from the posterior mesotympanum (Fig. 9.14). If the exposure is still restricted, a decision can be made intraoperatively to convert this to a canal wall down mastoidectomy. 1. Levenson MJ, Michaels L, Parisier SC. Congenital cholesteatomas of the middle ear in children: origin and management. Otolaryngol Clin North Am 1989;22 (5):941–954 2. Michaels L. Origin of congenital cholesteatoma from a normally occurring epidermoid rest in the developing middle ear. Int J Pediatr Otorhinolaryngol 1988;15 (1):51–65 3. Tos M. A new pathogenesis of mesotympanic (congenital) cholesteatoma. Laryngoscope 2000;110 (11):1890–1897 4. Derlacki EL, Clemis JD. Congenital cholesteatoma of the middle ear and mastoid. Ann Otol Rhinol Laryngol 1965;74 (3):706–727 5. Grundfast KM, Ahuja GS, Parisier SC, Culver SM. Delayed diagnosis and fate of congenital cholesteatoma (keratoma). Arch Otolaryngol Head Neck Surg 1995;121 (8):903–907 6. Litman RS, Smouha E, Sher WH, Shangold LM. Two cases of bilateral congenital cholesteatoma—usual and unusual presentations. Int J Pediatr Otorhinolaryngol 1996;36 (3):241–252
References
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


