Chapter 56 Coats Disease
History
Coats disease is an idiopathic condition characterized by telangiectatic and aneurysmal retinal vessels with intraretinal and subretinal exudation and fluid.1 Coats disease was first described by Scottish ophthalmologist George Coats in 1908.2 In his initial classification, Coats separated this new entity into three distinct groups. Group I included eyes with massive subretinal exudation and no demonstrable vascular abnormalities. Group II consisted of eyes with massive subretinal exudation and multiple retinal vascular abnormalities with intraretinal hemorrhage. Group III included eyes with massive subretinal exudation and frank retinal arteriovenous malformations.2 Eugen von Hippel later demonstrated that group III represented the distinctly separate entity of angiomatosis retinae (later retermed retinal capillary hemangioma or retinal hemangioblastoma), prompting Coats to drop this group from his classification. In 1912, and again in 1915, Theodor von Leber described a disease with similar telangiectatic and aneurysmal retinal vessels that lacked the massive subretinal exudation described by Coats.3,4 This condition was later named Leber multiple miliary aneurysms. In a 1916 paper, Leber concluded that what he had described was merely an earlier stage of the disease process identified by Coats.5,6 This conclusion was later reinforced by Reese, who described an eye with Leber miliary aneurysms that progressed into a classic case of Coats disease during long-term follow-up.7 Although some authors have disagreed, most authorities today classify Leber disease as an early or nonprogressive form of Coats disease.5,8,9
Histopathology, etiology, and pathogenesis
Early descriptions of Coats disease focused primarily on the morphology of the disease and attempted to explain the disease process solely on the basis of fundoscopic and histopathologic changes.10,11 The first observations published by Coats focused on histopathologic examination of six eyes enucleated for this condition that he received from various colleagues.2 From these histopathologic specimens, Coats described subretinal masses of fibrous tissue intimately adherent to the outer retina. Most of the eyes had associated retinal thickening, degeneration, and detachment, as well as diffuse vascular abnormalities. Four of the six eyes had associated retinal hemorrhages and most had cholesterol crystals. One eye had evidence of bone formation. Coats believed the vascular changes represented secondary manifestations of the underlying disease and that the exudation was secondary to organization and partial resorption of retinal hemorrhages, as he often found hemorrhages in association with the exudation.2 Coats also postulated that the primary process might be infectious in light of the mononuclear infiltrate found on histopathologic examination. This infectious theory of Coats disease was supported by a number of other reports, including that of Evens,12 Straub,13 and Müller,13 who suspected toxoplasmosis as the underlying cause. Subsequent investigations and the failure of anti-inflammatory adrenocorticotropic hormone and steroid therapies have refuted early theories of a primary infectious or inflammatory etiology.13,14
Pathologic samples of eyes with Coats disease were once abundant owing to enucleations for suspected intraocular tumor.15 Moreover, most histopathologic reports describe findings in eyes with advanced stages of the disease. Gross examination of enucleated eyes typically reveals bullous retinal detachment.16 Subretinal fluid has a viscous, lipid-rich, yellow appearance with glistening crystals.16 Microscopically, the subretinal exudation contains collections of foamy histiocytes and cholesterol clefts.16 The outer retina is thickened by exudation, with variable penetration of inner retinal layers.16 Areas of the inner retina also contain numerous, large, dilated, and telangiectatic vessels.16 Other vascular abnormalities include degenerated and hypocellular walls, narrowed lumen with or without thrombi, capillary dropout, neovascularization, as well as perivascular sheathing and/or pigmentation.1,9,17–23 Thickened vessel walls show characteristic heavy periodic acid–Schiff-positive deposits.1 Flat preparations prepared by trypsin digestion reveal vascular aneurysms measuring 50–350 µm, often forming large sausage-like outpouchings and located on shunt vessels.1 Advanced cases may have intraocular bone formation, with evidence of calcification on echography and computed tomography (CT) scanning that potentially confuses the diagnosis with retinoblastoma.24,25 Electron microscopy confirms diffuse structural abnormalities of the retinal vasculature. Tripathi and Ashton described prominent structural alterations of retinal vessels in areas of retinal thickening, with thickened walls mostly replaced by a laminated fibrous coating of basement membrane-like material.18 Most affected vessels were completely devoid of endothelium and pericytes, with red blood cells, plasma, and fibrin filling the lumina. There was patchy thinning or even absence of the vessel wall in some areas, with the vessel lumen extending to the basement membrane of adjacent glial cells.18 These findings support the widely held view that Coats disease is a disorder of vascular integrity.
Indeed, recent work in molecular genetics suggests that Coats disease may be part of a spectrum of related genetic disorders known as “retinal hypovasculopathies”26,27 which includes Norrie disease, familial exudative vitreoretinopathy (FEVR), fascioscapulohumeral muscular dystrophy (FSHD), and the osteoporosis pseudoglioma syndrome.27–35 These diseases share a similar ocular phenotype, characterized by a failure to vascularize fully the retinal periphery and associated telangiectatic, incompetent remnant vasculature. Each of these conditions can be related to abnormalities in the Wnt signalling pathway during retinal angiogenesis.26,27,36
Several reports have implicated a deficiency of Norrin, a retinal protein, in the pathogenesis of Coats disease.37,38 In one case report, a female with unilateral Coats disease gave birth to a son affected by Norrie disease.37 Both carried a missense mutation within the Norrie disease pseudoglioma (NDP) gene on chromosome Xp11.2. Further analysis on archival tissue from nine enucleated eyes from males with unilateral Coats disease revealed a mutation in the NDP gene in one subject. Knockout mouse models of Norrie disease model demonstrate abnormalities of the retinal vessels, including telangiectasia, bulb-like dilatations, and underdevelopment of the capillary bed.39,40 Elevated levels of hypoxia-inducible factor-1α and vascular endothelial growth factor (VEGF), as well as characteristic electroretinogram patterns in these mice, confirmed inner retinal hypoxia.40 Moreover, ectopic lens expression of Norrin in knockout mice induces the formation of normal deep retinal capillaries, and completely prevents abnormalities in retinal angiogenesis.41 These observations suggest a genetic basis for Coats disease and that a mutation in the NDP gene may be responsible.
Although FEVR and Coats disease share certain clinical features, they likely do not share a common genetic basis. Robataille and associates studied DNA expression of Frizzled-4 (FZD4) in 68 cases of FEVR and 16 cases of Coats disease.42 They found 11 FZD4 mutations in FEVR cases and no mutations in Coats disease cases. They concluded that germline mutations in FZD4 do not appear to be a common cause of Coats disease, implying that Coats disease does not likely represent asymmetric FEVR and is a distinct ocular condition.
Other genetic pathways have been explored. Cremers and associates found that 55% of eyes with retinitis pigmentosa and Coats-like secondary exudative vasculopathy contained a mutation in the CRB1 gene.43 This suggests that the CRB1 gene could be involved in primary Coats disease as well as other retinal diseases and dystrophies.
Clinical presentation
Coats disease is classically a painless ophthalmic condition. The disease affects males three times as often as females and has no reported racial or ethnic predilection.44 Coats disease is unilateral in 80–95% of cases.5,44,45 Some of the bilateral cases in older reports could represent secondary bilateral Coats-like retinopathy with systemic conditions and not true primary Coats disease. Any patient with bilateral presumed Coats disease should be evaluated for conditions that causes Coats-like exudative retinopathy such as retinitis pigmentosa, pars planitis, FSHD, FEVR, or other diseases.46 Based on a study of 150 consecutive cases, the mean age at diagnosis is 5 years with a range of 1 month to 63 years.44 Some speculate that the disease could be present at birth.13,47
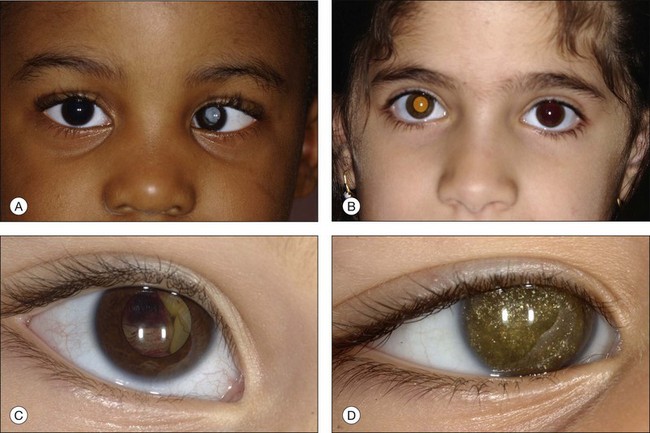
In an analysis of 150 consecutive cases of Coats disease, presenting symptoms included decreased visual acuity (43%), strabismus (23%: Fig. 56.1), leukocoria/xanthocoria (20%: Fig. 56.1), pain (3%), heterochromia (1%), nystagmus (1%), and no symptom (8%).44 Visual acuity was 20/20 to 20/50 in 12% of all eyes, 20/60 to 20/100 in 11%, 20/200 to counting fingers in 18%, and hand motions to no light perception in 58%. Nearly 90% of eyes had a normal anterior-segment examination. Those with findings included cataract (8%), iris neovascularization (8%), shallow anterior chamber (4%), corneal edema (3%), cholesterol in the anterior chamber (3%: Fig. 56.1),48,49 and megalocornea (2%). Retinal findings included telangiectasia (100%), intraretinal exudation (99%), exudative retinal detachment (81% with 42% demonstrating partial retinal detachment and 58% with total retinal detachment), retinal hemorrhage (13%), retinal macrocyst (11%), vasoproliferative tumor (6%), and optic disc neovascularization (1%). No eyes in this cohort presented with vitreous hemorrhage. Other studies have described macular fibrosis in up to 23% of patients, developing in an area of previous dense exudation, and involving the fovea in all cases.50
Adult cases of Coats disease are similar in clinical presentation and disease course, but generally have a smaller area of involvement, slower disease progression, more frequent hemorrhage near larger aneurysmal dilated vessels, and typically do not present with strabismus.51 Although the adult form of the disease has been described as frequently associated with hypercholesterolemia, such an association does not appear to occur in the juvenile form.14,52
The typical ophthalmoscopic picture in Coats disease is that of localized, yellow, subretinal exudation associated with adjacent vascular anomalies, including sheathing, telangiectasia, tortuosity, aneurysmal dilation, zones of capillary dropout, and occasionally neovascularization.13,44 There is variability in this clinical picture.44 Exudation, hemorrhage, or a combination appears minimal during less active stages of the disease and can progress to massive findings, obscuring the retinal vasculature in more aggressive stages. The vascular abnormalities can be subtle and clinically undetectable or can be obvious as a dominant feature. The clinical course is also variable, but is generally slowly progressive. Spontaneous remission has been observed but is exceptional.53 Subretinal choroidal neovascularization rarely occurs in areas of lipid deposition. As subretinal fluid and exudation increase, the retinal detachment progresses and can reach a highly elevated state, visible behind the lens (Fig. 56.1). Hemorrhagic and nonhemorrhagic retinal macrocysts can occur as a result of chronic coalescent intraretinal cystoid edema.54 Secondary complications such as iridocyclitis, cataract, and secondary neovascular glaucoma can lead to phthisis bulbi in severe cases.2,15,55
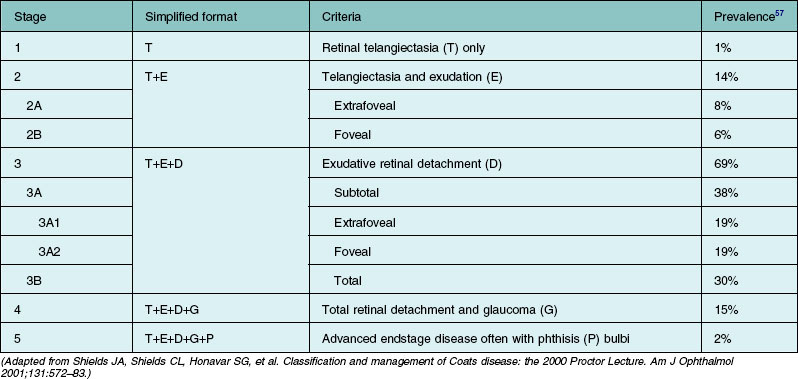
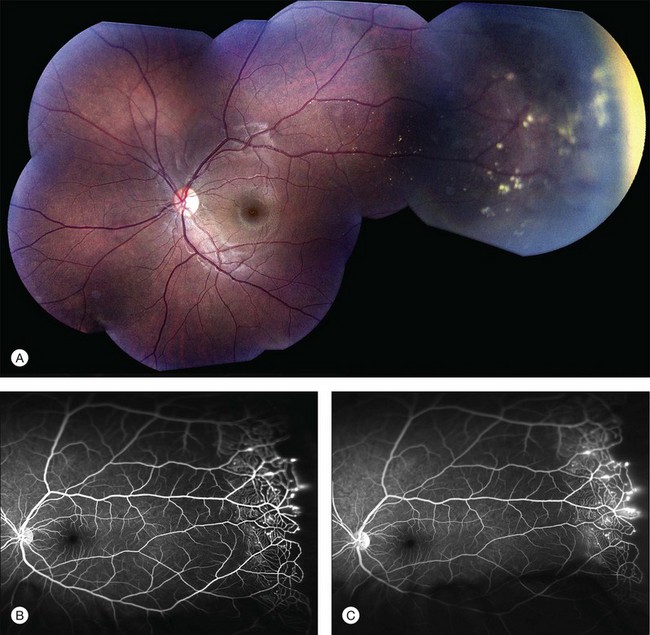
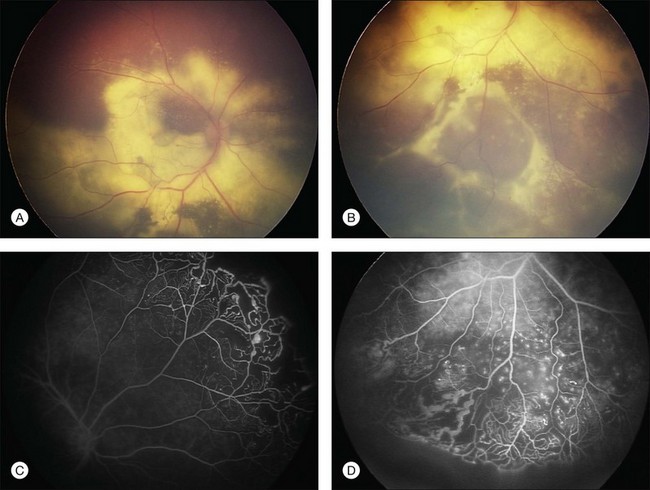
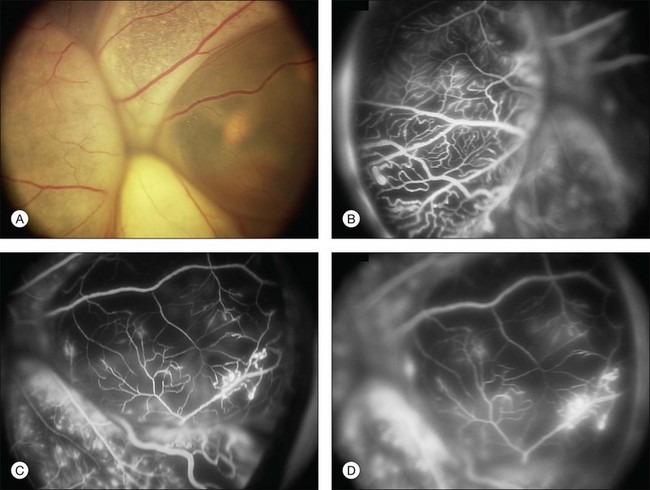
Several staging systems for Coats disease have been proposed.2,56 The most recent and widely used was introduced by Shields and associates in 2000 (Table 56.1).57 Stage 1 is characterized by retinal telangiectasia. Stage 2 has telangiectasia and intraretinal exudation, extrafoveal exudation defines stage 2A (Fig. 56.2), whereas foveal involvement defines stage 2B. Stage 3 is defined by exudative retinal detachment, 3A is a subtotal detachment, with 1 and 2 designating extrafoveal and foveal involvement, respectively (Fig. 56.3), and 3B is a total retinal detachment (Fig. 56.4). Stage 4 has total retinal detachment plus elevated intraocular pressure (Fig. 56.3), and stage 5 is endstage disease, occasionally with phthisis bulbi (Fig. 56.1). Using this classification for grouping of 124 cases, Shields and associates found stage 1 in 1%, stage 2 in 14% (2A in 8%, 2B in 6%), stage 3 in 69% (3A1 in 19%, 3A2 in 19%, 3B in 30%), stage 4 in 15%, and stage 5 in 2%.57 Based on classification and treatment, visual acuity strongly depended on stage at diagnosis.44,58 At the time of Coats disease control, poor visual acuity (20/200 or worse) was found in 0% of stage 1, 53% of stage 2, 74% of stage 3, and 100% of stage 4 and stage 5.57
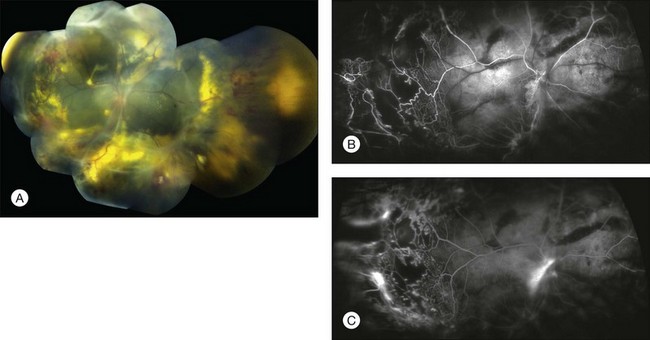
Diagnostic testing
Ancillary testing may be useful when the diagnosis of Coats disease is suspected, but other clinical entities must be ruled out, most notably retinoblastoma.59 Particularly when retinal detachment with subretinal exudation and dilated retinal vessels coexist, even an experienced clinician may have difficulty differentiating these entities ophthalmoscopically. Fluorescein angiography is critical to document classic findings to establish the diagnosis. Echography may enable differentiation between Coats disease and retinoblastoma on the basis of features such as the character of the retinal detachment and the presence or absence of subretinal calcifications. Echography is less useful when the retinoblastoma is poorly calcified and also has shortcomings in detecting optic nerve or extraocular extension of retinoblastoma when heavy calcification exists.60
Fluorescein angiography
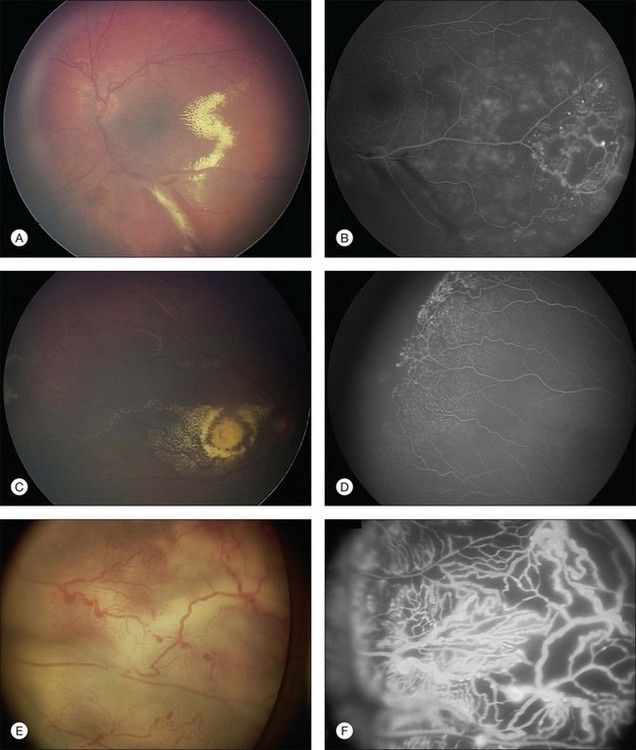
The typical fluorescein angiographic picture of Coats disease is one of numerous localized anomalies of the retinal vasculature with peripheral retinal nonperfusion (Figs 56.2–56.6). Telangiectasia, aneurysms, beading of vessel walls, and various vascular communicating channels are found with larger vessels. These vessels show early and persistent leakage, which verifies their role as the source of exudation and hemorrhage. This leakage represents breakdown of the blood–retinal barrier, which can further be detected by vitreous fluorophotometry.61 Microvascular involvement is demonstrated with areas of diffuse loss of the capillary bed or areas of complete capillary nonperfusion on angiography. Geographic zones of microvascular involvement are typically surrounded by areas of arteriolar and venular anomalies. Although some early authors described cases of massive exudation or hemorrhage without areas of obvious vascular involvement, fluorescein angiography and histopathologic specimens invariably show unsuspected anomalous vessels. The ability to identify all anomalous vessels, especially those with the greatest degree of leakage, has made adequate treatment of Coats disease a possibility.5,57,62,63
Computed tomography
CT is valuable because of its ability to characterize intraocular morphology, quantify subretinal densities, identify vascularities within the subretinal space through the use of contrast enhancement, and detect other abnormalities that may be associated in the orbital or intracranial space. Spiral CT has the advantage of reducing anesthesia risk in small children and also decreases acquisition time and staff and equipment monitoring requirements.64
Magnetic resonance imaging
Magnetic resonance imaging (MRI) as an auxiliary test is useful because it permits multiplanar imaging and superior contrast resolution and yields biochemical insight into the structure and composition of tissues.65,66 It is less useful in detecting calcium than either ultrasound or CT scanning.60,67 An MRI study of 28 patients with leukocoria or intraocular mass, or both, found that retinoblastomas could be reliably distinguished from Coats disease, toxocariasis, and persistent hyperplastic primary vitreous.67 Calcification could not be reliably detected on MRI scanning.
Doppler ultrasonography
High-resolution Doppler ultrasound has been suggested as a diagnostic adjunct, providing unique information with real-time imaging and duplex pulse Doppler evaluation.68 This technique may delineate structural abnormalities not shown by CT or MRI.
Blood testing
Aqueous lactic dehydrogenase and isoenzyme levels have not proved valuable in distinguishing between Coats disease and retinoblastoma. Examination of subretinal fluid, although rarely used, is accurate in confirming the diagnosis of Coats disease on the basis of cholesterol crystal and pigment-laden macrophages in the absence of tumor cells.60
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


