Chromosomes: Structure, Function, and Clinical Syndromes
Brian J.R. Forbes
Leonard B. Nelson
Sanger’s demonstration in 1953 that proteins are unique linear arrays of amino acids and the suggestion by Watson and Crick in that same year that DNA, the genetic material, is similarly a linear array of bases gradually led to the realization that genes are expressed by the linear translation of the base sequence of nucleic acids into the amino acid sequence of proteins. The genetic code is simply the sequence relationship between bases in genes and the amino acids they encode. The entire genetic blueprint of humans is composed of linear double-helix deoxyribonucleic acid (DNA) and is present in the 46 chromosomes of every human nucleated cell. Each species has a characteristic number of chromosomes. The functional unit of the genetic blueprint is the gene, a segment of DNA required to code for one polypeptide. The human genome was previously believed to contain approximately 100,000 distinct genes, however, the Human Genome Project reported that value as being closer to 30,000.1
The disorders discussed in this chapter are the result of loss, duplication, or rearrangement of whole groups of adjacent genes. As a result, patients show generalized and widespread systemic abnormalities, depending on the number and relative importance of the genes lost or duplicated. The chromosomal disorders may involve the entire genome resulting in triploidy or polyploidy or the disorders may be numerical resulting in monosomies and trisomies. Even with the latest banding techniques, the smallest detectable deletion or duplication of a single chromosome contains many genes.
The common dominant, recessive, or X-linked genetic disorders are the result of single-gene defects. Although generalized abnormalities are frequently present, they are caused by the effect of that single-gene product on many different aspects of development. Detection requires molecular techniques because the single-gene defects are not visible by cytogenetic techniques. Chromosomal disorders may be inherited in a dominant manner, but their generalized abnormalities are the result of multiple-gene imbalances and their generalized defects are often so severe that affected individuals often do not reproduce.
GENE DOSE EFFECTS IN CHROMOSOMAL ABNORMALITIES
The 46 chromosomes exist in 23 homologous pairs in the normal individual; one chromosome of each pair is from the mother, the other is from the father and one of each pair is transmitted to each child. Twenty-two pairs of chromosomes are alike in males and females and are, hence, called the autosomes; the two sex chromosomes, the remaining pair, differ in males and females. The members of the homologous pair carry the same genetic information; they have the same gene loci in the same sequence, although at any one locus they may have either the same or different alleles. Each of the human genes is, therefore, paired, and the total expression of any particular gene is the sum of the products coded for by both the maternal and paternal copies, or alleles, of that gene. Chromosomal abnormalities will, as a rule, involve only one of the two chromosomes in any pair. When a chromosome or part of chromosome is missing, the impaired alleles on the other chromosomes are expressed without modification. In the case of genes that code for enzymatic proteins, total activity will be reduced by 50% if the enzyme activity is gene dose dependent. On the other hand, when an entire chromosome or a position of one is duplicated, the genes in that segment have three alleles, and the gene product is present in 150% of normal amounts.
The abnormal dose effect of multiple genes in chromosomal disorders may have widespread effects on the developing organism. In Drosophila, mutations or deletions involving as many as 41 loci on all four chromosomes produced a similar phenotype consisting of slow development, small body size, large rough eyes, thin wings, and low fertility. These 41 loci represent sites of synthesis of transfer DNA, deficiency of which results in a generalized decreased rate of protein synthesis.2 Many of the human chromosomal abnormalities share common features: mental retardation, low fertility, growth retardation, and coarse and abnormal facial features, among others. In humans, as in Drosophila, the recurrent and overlapping phenotypes seen with many of the chromosomal abnormalities can be explained on the basis of a generalized decreased rate of protein synthesis.
MECHANISMS IN OCULAR CHROMOSOMAL DISEASE
Table 1, which summarizes the ocular features of some of the partial trisomies and partial deletion chromosomal syndromes, demonstrates the considerable phenotypic overlap among the various chromosomal disorders. It is believed that abnormal rates of cell division at critical times in embryogenesis, likely the result of abnormal gene dose effect, result in the generalized growth retardation and the congenital anomalies noted at birth.2 For example, closure of the fetal choroidal fissure takes place between the fifth and seventh week of gestation. If an embryo has an abnormal karyotype that causes retardation of localized cell division, the result may be that closure of the fetal choroidal fissure will not occur. Uveal coloboma is a feature of chromosomal disorders involving many different chromosomes.
TABLE 1. Ocular Signs in Partial Trisomy and Partial Deletion Chromosomal Syndromes | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Many of the ocular abnormalities noted in the chromosomal disorders are not chromosome specific; rather, they are secondary phenotypes. Orbital hypotelorism is found in many chromosomal disorders. In trisomy 21, the abnormally narrow space between the orbits is a result of primary hypoplasia of the central facial structures, whereas in the 13q+ and the 18p- syndromes the hypotelorism is associated with some degree of holoprosencephaly and trigonocephaly. In both situations, however, the end result is a phenotypic anomaly that results secondarily from a more basic defect in development as well. The ocular pathology in embryos with chromosomal abnormalities demonstrated that not only does retardation of development occur but also that various forms of dysgenesis of ocular structures occur. Specific findings included microphthalmia, cataract, subluxation of the lens, staphyloma of the optic nerve, retinal dysplasia, and cyclopia.3
Microphthalmia is an ocular abnormality seen in several chromosomal syndromes. The microphthalmia in trisomy 13 is probably related to the presence of holoprosencephaly. However, because microphthalmia is seen in other syndromes in which holoprosencephaly is not present, the defect must be nonspecific or related to some other specific gene locus. It is known, for example, that mutations on all four chromosomes of Drosophilia may produce small eyes or anophthalmia. In the mouse, genes on three separate chromosomes may result in the same ocular anomalies. Deletion or duplication of different portions of the human karyotype can result in microphthalmia. Hypertelorism, antimongoloid lid slant, epicanthal folds, highly arched eyebrows, strabismus, and colobomas are abnormalities that are frequently found among the chromosomal disorders (see Table 1).
A HISTORY OF HUMAN CYTOGENETICS
Before 1956 there were believed to be 48 human chromosomes; however, in that year Tjio and Levan4 showed that 46 was the correct number. In 1979, Hsu published a book reviewing the history of human cytogenetics in which he divided the subject into four eras: the dark ages before 1952, the hypotonic period from 1952 to 1958, the trisomy period between 1959 and 1969, and the current chromosome banding era that started in 1970.
In the dark ages, Painter, in 1923, was studying slides of paraffin-sectioned testes and suggested 48 as the number of chromosomes in humans. Because his paper was cited repeatedly that number was eventually repeated as fact. During this era, the squash technique for separating human metaphase chromosomes was developed, and the chemical prefixation technique and the use of colchicine to block cell division metaphase were also introduced. In 1952, Hsu introduced the hypotonic technique, which leads to control of swelling of the cells and separation of individual chromosomes. It was this technique that allowed Tjio and Levan to determine the correct number of human chromosomes. The techniques introduced during the hypotonic era were applied to the analysis of chromosomes from individuals who were mentally retarded or who had other congenital anomalies. In 1959, Lejeune and associates described the first known human trisomy, Down syndrome. In that same year, Ford and associates reported that patients with Turner’s syndrome had only one of the two X chromosomes, for a total of 45 chromosomes instead of 46. Rapidly thereafter, additional chromosomal defects were defined, and the cataloguing of human trisomies compatible with life was exhausted. Attention was then turned to structural aberrations and their phenotypic consequences.
Most human chromosomes could not be identified individually before 1970. In that year, Caspersson and associates5 applied fluorescence microscopy to human chromosomes. It was found that chromosomes, when stained with quinacrine, exhibited fluorescent cross bands of different widths. Subsequently, nonfluorescent banding was achieved through a number of different techniques, including various ways of partially denaturing chromosomal DNA. With several of these partial denaturation techniques, subsequent Giemsa staining resulted in reproducible banding patterns that were unique for each chromosome. The technique of banding prophase chromosomes made it possible to define chromosomal segments and break points even more accurately. Through banding techniques, small deletions or duplications of segments of DNA within the chromosome can be precisely identified. This advance made possible the description of a whole new class of chromosomal diseases, the partial trisomy and partial deletion syndromes.
INCIDENCE OF CHROMOSOMAL ABNORMALITIES
CHROMOSOMAL SYNDROMES
The abbreviations and terminology commonly used in describing chromosomal rearrangement are as follows. The letter p is used to represent the short arm of a chromosome and the letter q the long arm. The letters are used after the number of the chromosome under discussion. A plus sign (+) after the letter indicates duplication or partial trisomy for the portion of the arm indicated. A minus sign (-) indicates a partial deletion for a portion of that arm. For example, the 5q+ syndrome is the name used to refer to the clinical entity resulting from any partial duplication of the long arm of chromosome 5. More precise localization along a chromosome arm is provided by a numbering system based on prominent bands beginning at the centromere. Each arm of a chromosome is divided into three to five regions, and these are again subdivided into three or more subunits. Using this system, a single band can be named.
THE TRISOMY SYNDROMES
Trisomy or monosomy for autosomes other than 13, 18, 21, and 22 are almost invariably lethal and are frequently seen in spontaneous abortions.
Trisomy 13 (Patau Syndrome)
Trisomy 13 occurs among newborns at a frequency of 1 in 20,000.11 High maternal age is a factor predisposing to trisomy 13, as it is for other trisomies. About 45% of affected infants die before the age of 1 month with only 5% reaching the age of 3 years, and there are only 5 reports of patients surviving beyond the first decade.12 Wide phenotypic variability in trisomy 13 is the rule and may be secondary to the variable allelic content of the three homologous chromosomes. Partial trisomy for different parts of chromosome 13 has been described. Patients with these partial trisomies have only some of the features of trisomy 13. A clinical chromosomal map has been constructed by matching common clinical features with common regions of chromosome involvement. Clinical features of trisomy 13 include severe mental retardation resulting from holoprosencephaly, cleft lip and palate, and polydactyly (Fig. 1). These patients are often deaf and may have capillary hemangiomas and scalp defects (Fig. 2). The heel is often prominent and the bottom of the foot has the shape of the runners on a rocking chair.13 The ocular features in trisomy 13 range from anophthalmia to microphthalmia. A fleshy pterygium-like corneal opacification may be seen (Fig. 3), and iris and choroidal colobomas are common (Fig. 4). Hypertelorism occurs in almost all cases.14
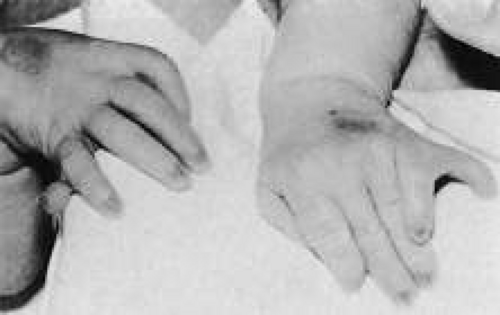 Fig. 1. Postaxial polydactyly in a patient with trisomy 13. A skin tag is present on the right hand. (Courtesy of Irene Maumenee, MD) |
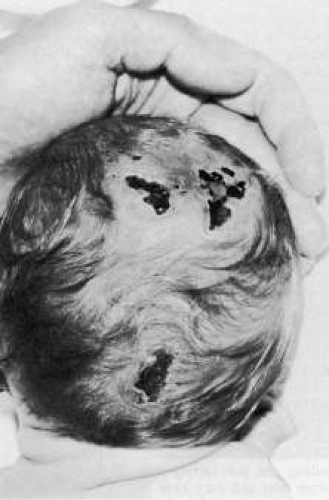 Fig. 2. Scalp defects in a patient with trisomy 13. (Courtesy of Irene Maumenee, MD) |
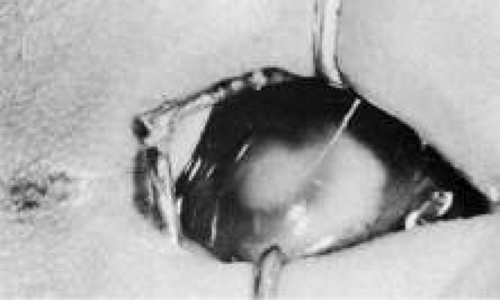 Fig. 3. Central corneal opacity in a patient with trisomy 13. |
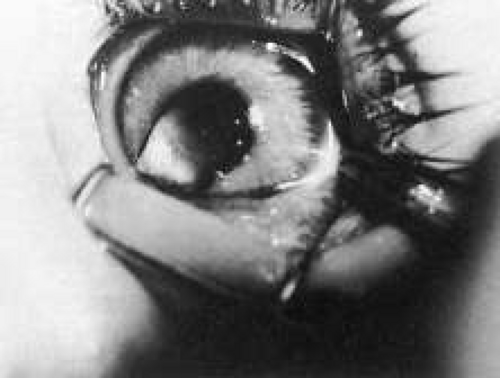 Fig. 4. Inferior nasal coloboma and a corresponding sector cataract in a patient with trisomy 13, who is probably mosaic. The lens opacity had been stable for 2½ years. |
Trisomy 18 (Edward’s Syndrome)
Trisomy 18 occurs among newborns at a frequency of 1 in 6000.15 High maternal age is a factor predisposing to trisomy 18, and about 90% of affected infants die before the age of 6 months with only about 5% reaching the age of 1 year. A nearly diagnostic feature for this syndrome is the positioning of the thumb and fingers in a tightly clenched fist. The thumb is flexed inside the fist with the index finger overriding the third digit. Infants with trisomy 18 have a characteristic constellation of clinical features that includes mental retardation, hypertonicity, cardiac defects, low-set malformed ears, and flexion deformities.16 Ocular malformations are commonly found in trisomy 18 patients with hypertelorism described in 87% of patients, and prominent epicanthal folds, microphthalmia/anophthalmia, and ptosis seen in about one third of cases. Other less commonly described findings include globe abnormalities such as corneal opacity, anterior segment abnormalities, cataract, and optic nerve pits and colobomas.17
Trisomy 21 (Down Syndrome)
Down syndrome, the most frequent form of mental retardation caused by a microscopically demonstrable chromosomal aberration, is characterized by well-defined and distinctive phenotypic features and natural history; Down syndrome occurs in 1/800 newborns.18 Trisomy 21 is the most common of all the trisomy syndromes and is the result of aneuploidy involving the smallest human chromosome; 75% of these embryos are aborted spontaneously. Incidence increases with maternal age; the risk of having a liveborn with Down syndrome at maternal age 30 is 1 in 1000 and at maternal age 40 is 9 in 1000.19
The characteristic face of a child with trisomy 21 exhibits epicanthus, oblique palpebral fissures, a protruding tongue, a flat nasal bridge, and low malformed ears. Individuals with Down syndrome have specific major congenital malformations (found in 30% to 40% of patients) such as those involving the heart, particularly the atrioventricular canal, and the gastrointestinal tract. Patients with Down syndrome also have between 10 to 20 times greater risk of leukemia than the chromosomal normal population. In particular, there is an increase of 200 to 400 times the incidence of acute megakaryocytic leukemia. The affected child is mentally retarded and has shortened anteroposterior (AP) skull diameter; short stature; a typical dermatoglyphic pattern that consists of a simian line, distal palmar axial triradius, and a high frequency of ulnar loops; and a life expectancy that is reduced to 71% at 30 years. In the most common form of trisomy 21, there are three free copies of chromosome 21; in about 5% of patients one copy is translocated to another acrocentric chromosome, most often chromosome 14 or 21.20
Eye findings can be extensive and include Brushfield spots, which are circumferentially distributed white spots on the iris. Brushfield spots are a hallmark of Down syndrome and are found in one third of patients. Similar lesions may be present in the chromosomally normal individuals in whom they are referred to as Wofflin nodules. Lens changes are present in the Down syndrome population with approximately 300-fold greater incidence than the general population (11%). Epicanthal folds with short or sparse eyelashes are present in more than 50% of patients. Other eye abnormalities include strabismus (57%), esotropia (90%) greater than exotropia (5%), and frequently associated A or V patterns. Extremes in refractive errors are prevalent in the form of myopia (23%), hyperopia (21%), and astigmatism (22%). Nystagmus has a reported incidence of 29% without specified type. Keratoconus develops in some 15% of Down syndrome patients, and nasolacrimal duct obstruction is present in 15% of patients.21
Trisomy 22
Trisomy 22 is a rare disorder with only 20 reported cases. It is lethal in the immediate postpartum period. Mean maternal age at the time of birth is 30.5 years.22 All of the patients have obvious mental and physical retardation. The most common congenital abnormalities include microcephaly, preauricular skin tags, congenital heart disease, micrognathia, a long philtrum, malformed ears, and a long nose. At least 50% of the patients had cleft palate, malposed thumb and long slender fingers, congenitally dislocated hips, and hypotonia. As many as 30% have hypoplastic or lowset nipples. Cryptorchism was noted in all males. The most common ocular malformations are chorioretinal colobomas and microphthalmia. Other less common features include strabismus and antimongoloid lid slant.23
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


