Chapter 41 Vitreous
Introduction
The development of the vitreous body and zonule can be divided into three stages:
1. The primary vitreous is formed during the first month of gestation and is a vascularized mesodermal tissue separating the developing lens vesicle and the neuroectoderm of the optic cup. It contains branches of the hyaloid artery that later regress.
2. The secondary vitreous starts at 9 weeks and develops throughout embryonic life. It forms the vitreous body, is avascular and transparent, and displaces the primary vitreous, which becomes Cloquet’s canal, running from the optic disc to the lens. By the third month, the secondary vitreous fills most of the developing vitreous cavity.
3. The tertiary vitreous lies between the ciliary body and lens, separated from the secondary vitreous as well-formed fibrils, which later develop into the zonule.
Developmental anomalies of the vitreous
Vitreous cysts
Acquired cysts occur with inflammatory disease and, rarely, with juvenile X-linked retinoschisis.
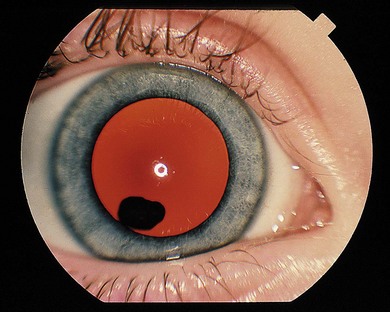
Cysts may lie in the vitreous immediately behind the lens (Fig. 41.1) or in the posterior vitreous. They may be mobile or attached to the lens or optic disc. Mostly, intervention is not required; occasionally laser treatment (Nd : YAG or argon laser) may be used to collapse the cyst if it is symptomatic. However, repeat Nd : YAG therapy of an anterior pigmented cyst has resulted in a cataract.1
Persistent fetal vasculature (persistent hyperplastic primary vitreous) (see Chapter 35)
Persistent fetal vasculature (PFV) persistent hyperplastic primary vitreous (PHPV) is caused by failure of the primary vitreous to regress. Most cases are sporadic and unilateral; there may be minor abnormalities in the fellow eye. Bilateral and familial cases have been reported but these are likely to represent cases of vitreoretinal dysplasia (see Vitreoretinal dysplasia).
For anterior PFV, see Chapter 35.
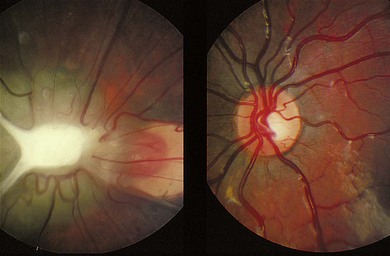
In posterior PHPV, the ocular abnormality is confined to the posterior segment, and may present with leukocoria, strabismus, microphthalmia, or nystagmus. The lens is usually clear. There is often a fold of condensed vitreous and retina running from the optic disc to the ora serrata or lens (Fig. 41.2), often associated with a retinal detachment. Ultrasound and CT imaging help in differentiating PHPV from retinoblastoma (see Chapter 42).
Vitreoretinal dysplasia
Maldevelopment of the vitreous and retina, vitreoretinal dysplasia, is seen either as an isolated abnormality or associated with systemic abnormalities.2 Syndromes such as Norrie’s disease and Warburg’s syndrome may have bilateral vitreoretinal dysplasia. It also occurs in trisomy 13, trisomy 18, triploidy, and in association with cerebral malformations.
Norrie’s disease
Clinical and histologic findings
Norrie’s disease is an X-linked recessive disorder; affected males are blind at birth or in early infancy. Twenty-five percent of affected males are developmentally delayed; one-third later develop cochlear deafness. A more severe systemic phenotype is seen in patients with large chromosomal deletions which encompass the Norrie’s gene locus. Ocular findings include bilateral cataracts, bilateral retinal folds, retinal detachment, vitreous hemorrhage, and bilateral vitreoretinal dysplasia (Fig. 41.3). The retinal detachments are usually of early onset and have been observed in utero by ultrasonography. Most cases progress to an extensive vitreoretinal mass and bilateral blindness.
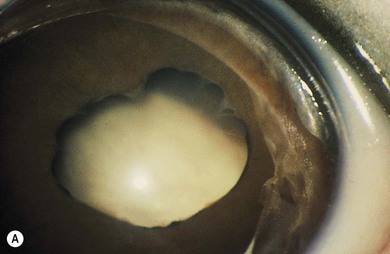
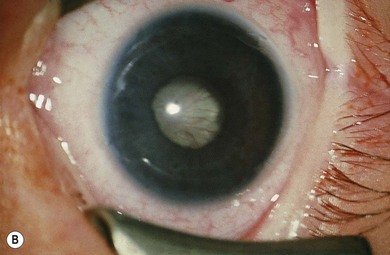

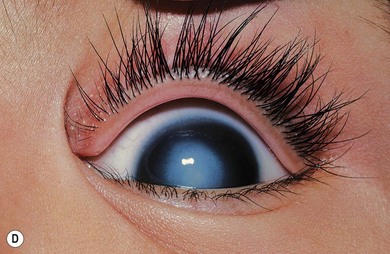
Angle closure glaucoma may develop; this is best managed by lensectomy. Late signs include corneal opacification, band keratopathy, and phthisis bulbi (see Fig. 41.3).
Carrier females do not usually show any ocular abnormality and electroretinography (ERG) is normal. An affected female has been reported, born to a carrier mother, who had a retrolental mass in the right eye and a retinal fold with a tractional retinal detachment in the left. Molecular genetic testing confirmed that she was a manifesting heterozygote. She showed skewed X-inactivation in her peripheral blood lymphocytes suggesting non-random inactivation, with inactivation occurring more frequently in the normal rather than the mutant X chromosome. A female with Norrie’s disease, with an X autosome translocation has also been described.3
Molecular genetics and pathogenesis
More than 100 mutations have been identified in the Norrie’s disease gene, NDP. The gene is expressed in the neural layers of the retina, throughout the brain, and in the spiral ganglion and stria vascularis of the cochlea. The encoded protein, Norrin, is a component of the Wnt signaling pathway, a key regulator of various stages of ocular development, including retinal field establishment, maintenance of retinal stem cells, vasculogenesis in the retina, formation of the ciliary body, and cornea and lens development.4 Wnt-signaling is important in retinal development: uncontrolled, it may cause other retinal diseases including familial exudative vitroretinopathy, the osteoporosis-pseudoglioma syndrome, and Norrie’s disease. The association of Norrie’s disease with peripheral vascular disease is evidence for a role of the Norrin gene in extraocular angiogenesis.
The identification of the Norrie’s disease gene allows molecular genetic diagnosis of the carrier state and prenatal diagnosis. Norrie’s disease may be associated with chromosomal deletions involving the NDP locus, the adjacent monoamine oxidase genes MAOA and MAOB, and additional genetic material. Children with such deletions have a more severe (“atypical”) phenotype.5 In addition to the characteristic retinal dysplasia, the “atypical” phenotypes may include: severe learning difficulties, involuntary movements, atonic seizures, hypertensive crises, and hypogonadism.5
Coats’ disease may be caused by somatic mutations in NDP only in the retina of affected eyes; the somatic mutations result in deficiency of Norrin with consequent abnormal retinal vascular development, the hallmark of Coats’ disease.6
Trisomy 13
Clinical and histologic findings
Bilateral ocular abnormalities are seen in almost all cases of trisomy 13; the common ocular findings are detailed in Box 41.1. Affected infants often show total disorganization of the vitreous and retina. Histology shows extensive retinal dysplasia. Intraocular cartilage is frequent and may be characteristic.
Incontinentia pigmenti (Bloch-Sulzberger syndrome)
Clinical and histologic findings
Incontinentia pigmenti (IP) is an uncommon X-linked dominant disorder affecting the skin, bones, teeth, central nervous system, and eyes, which is usually lethal in utero in males, leading to a marked female preponderance. The characteristic skin lesions appear soon after birth with a linear eruption of bullae predominantly affecting the extremities (Fig. 41.4A). The bullae gradually resolve to leave a linear pattern of pigmentation (Fig. 41.4B).
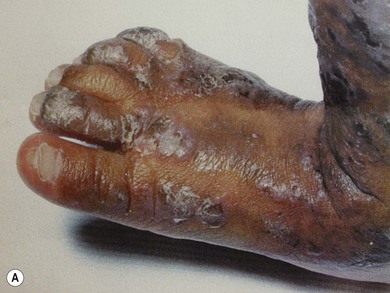
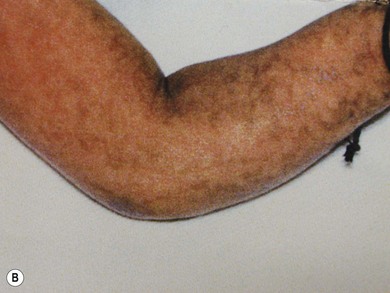
Ocular abnormalities are common including amblyopia, strabismus, nystagmus, optic atrophy, and retinal changes.7 The retinal changes are the cause of severe visual impairment that may be seen in IP. Corneal abnormalities include whorl-like epithelial keratitis with epithelial microcysts, mild mid-stromal “haziness,” and corneal subepithelial anterior stromal opacities.
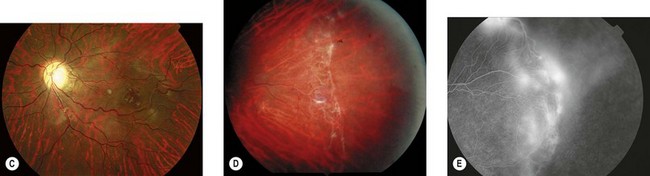
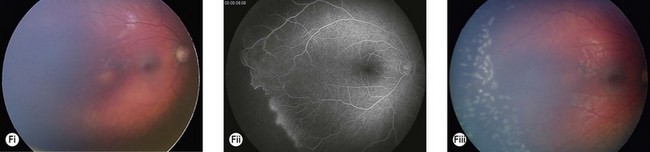
The most serious complication is retinal detachment, which may lead to severe visual impairment. Retinovascular abnormalities are common and include retinal vascular tortuosity, capillary closure, and peripheral arteriovenous shunts (Fig. 41.4C–E). These are most marked in the temporal periphery and may be associated with retinal neovascularization. Fluorescein angiography demonstrates areas of non-perfusion in the temporal periphery (Fig. 41.4E). Retinal neovascularization, if left untreated, may lead to tractional retinal detachment.
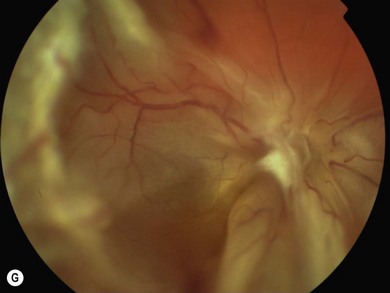
An ophthalmologist should regularly assess affected females soon after diagnosis and during infancy, in order to detect those cases of retinal vascular non-perfusion requiring treatment, usually occurring in the first 2 to 3 years of life. We undertake a retinal examination in affected infants every 2 months for the first year of life then, if normal, 4 monthly for the next 12 months then 6 monthly. If any peripheral retinal abnormality is seen or if a view of the retinal periphery is not obtained after repeated examination, examination under anesthetic is indicated. We combine this with RETCAM fluorescein angiography to document peripheral retinal non-perfusion and neovascularization. Laser treatment is indicated when there is significant capillary non-perfusion (Fig. 41.4F). Established retinal detachment is difficult to manage and has a poor visual outcome (Fig. 41.4G). It is rarely bilateral.
Molecular genetics and pathogenesis
IP is caused by mutations in the ubiquitously expressed gene, NEMO (NF-κB essential modulator). The NEMO protein is the regulatory component of the IκB kinase (IKK) complex, a central activator of the NF-κB transcriptional signaling pathway.8 In IP, loss-of-function mutations in NEMO lead to a susceptibility to cellular apoptosis in response to TNF-α.8
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


