Chapter 23 Neurogenic tumors
Optic nerve tumors
Optic glioma
Juvenile gliomas (World Health Organization Grade I pilocytic astrocytomas) consist of a surplus of the astrocytic glial cells supporting axons. They are the most frequent intracranial tumor to affect the visual pathways in children. They may occur anywhere in the brain, however, and are present in approximately 25% of young children with neurofibromatosis type 1 (NF1),1 itself the most commonly identified pathogenetic mutation in the general population with an incidence of approximately 1/3000. Most of these lesions are detectable by magnetic resonance imaging (MRI) in infancy, but only a fraction ever cause clinical symptoms. A roughly equivalent number of patients also present clinically with juvenile optic gliomas, but do not have NF1. Recent evidence reaffirms earlier notions that these tumors represent glial cell hamartomas.2,3 Grade II and higher malignant gliomas of the anterior visual pathway that behave more aggressively, including glioblastoma multiforme, are recognized in adults4,5 and may occasionally occur in children;6–9 their diagnosis and treatment, altogether different, is not addressed here.
Visual pathway gliomas
Gliomas may arise anywhere along the visual pathways. Multifocality is common, and an initial optic nerve presentation later followed by chiasmal involvement should not be assumed to necessarily represent invasion.10 Multifocal growth, as well as regression, may occur contemporaneously or at different times with either or both optic nerves affected, sparing chiasm, or vice versa (Fig. 23.1).11 Hence surgical resection of a distal optic nerve tumor may not necessarily prevent later growth more proximally.10,12 Patients with optic gliomas should be examined for signs of neurofibromatosis in the eye (in particular melanocytic iris hamartomas termed Sakurai-Lisch nodules), orbit (neurofibromas, sphenoid bone defects), and skin (multiple café-au-lait spots, neurofibromas). Family members should also be examined.
Though some have believed that gliomas occurring in the presence of NF1 have a better visual prognosis compared to those occurring sporadically, such impressions are due to selection biases; since most NF1-associated gliomas are detected by surveillance MRI scans ordered for asymptomatic NF1 patients, many clinically insignificant tumors are thus detected. Individuals without NF1, on the other hand, present to the physician only when they have large, symptomatic masses. Overall survival for those with NF1-associated gliomas, moreover, is worse; since these patients have an underlying mutation in the NF1 tumor suppressor gene, they tend to develop, for reasons not well understood,13 other, non-neural crest derived, tumors, particularly soft-tissue sarcomas,14,15 and myelogenous disorders.
Presentation
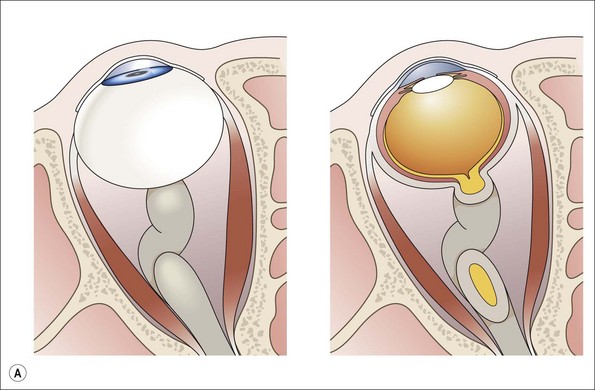
The age of presentation ranges from birth to between 4 and 12 years generally; the vast majority have presented by 20 years of age.16 Gliomas involving the intraorbital portion of the nerve may present with axial proptosis12 (Fig. 23.2). For reasons yet unclear, the enlarged intraorbital portion of the tumor has a predisposition to kink and deflect downward, thus producing an upward rotation of the posterior aspect of the globe (Fig. 23.3A,B): this can produce a nearly pathognomonic feature of mild proptosis in a child with hypotropia or limitation of elevation.12 Older patients may verbalize visual loss with color vision and field defects.
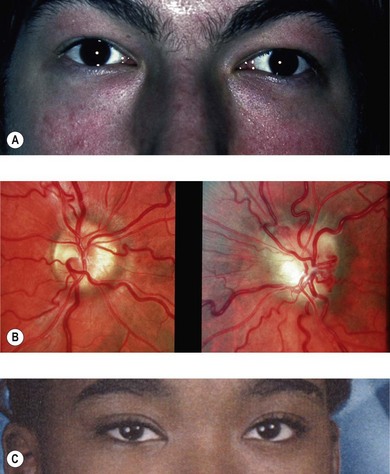
Chiasmal gliomas may present slightly later with bilateral visual loss, although they may also be discovered during investigation of hydrocephalus, disconjugate nystagmus (see Fig. 23.1), endocrine dysfunction,2 or in a hitherto asymptomatic patient via neuroimaging studies (Fig. 23.4).17 The presenting history is usually of slowly deteriorating vision, though in rare cases sudden visual loss can result from hemorrhage within the tumor.18,19
Gliomas affecting the chiasm and the nerves asymmetrically may cause a dissociated nystagmus mimicking spasmus nutans.20 In children, asymmetric nystagmus, particularly in the presence of optic atrophy, poor feeding, or hydrocephalus, is suggestive of chiasmal glioma with or without posterior extension into the optic tracts (see Fig. 23.1).
Involvement of the hypothalamus, whether primary or secondary, can produce various endocrine abnormalities. Precocious puberty is often present in children with chiasmal glioma.17 Reduced growth and sexual maturation, diabetes insipidus, and obesity may also occur. In infancy, there may be extreme wasting, reduced development, and often the vertical or asymmetric rotary nystagmus mimicking spasmus nutans, an association known as Russell diencephalic syndrome.6,21
Optic disk pallor may be noted if there is poor vision. Optociliary shunt vessels are present only if the tumor is in proximity to the globe with swelling of the optic disk compressing the central retinal vein. However, many of these overgrowths of glial cells which are intrinsic to the visual pathway do not significantly affect vision and remain clinically silent (see Fig. 23.4). Visual acuity does not correlate to tumor size or growth; vision can remain unaffected by large uniform tumor involvement along much of the visual pathway, or be extremely compromised by a small irregular growth, or regression, distorting axons focally.22 Hence, findings from visual evoked potential testing are generally not of use. In the absence of visual symptoms, visual pathway gliomas are rarely identified by fundus examination alone.
Radiographic features
Since the hamartomatous overgrowth of the astrocytic glial cells is intrinsic to the visual pathways, the usual contours of involved structures are preserved. As a result, the radiographic appearances are generally so characteristic that biopsy is not needed to make a diagnosis. Biopsy, moreover, carries a risk of ocular or visual morbidity and is frequently misleading in optic nerve glioma since there may be reactive changes in the surrounding arachnoid similar to, and misinterpreted as, nerve sheath meningioma.23,24
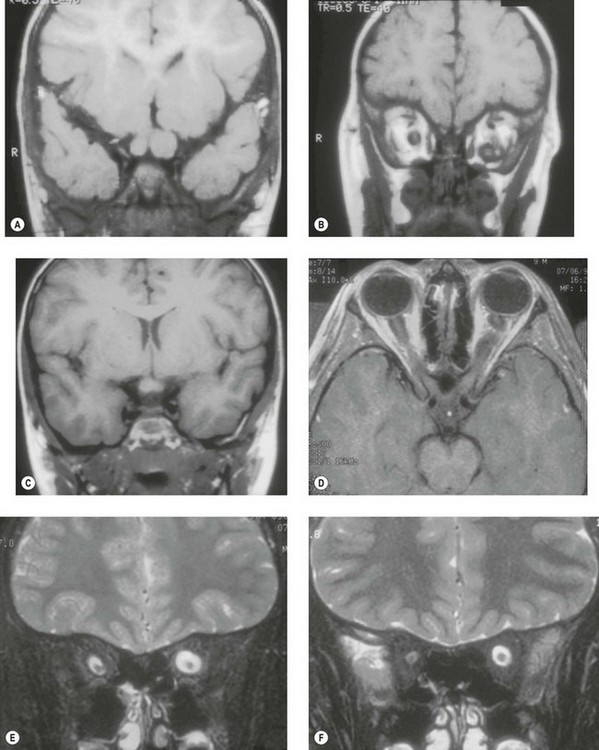
MRI (Figs 23.1, 23.4, and 23.5) is thus the modality of choice for evaluation and follow-up. MRI reveals a smooth fusiform optic nerve enlargement with variable gadolinium contrast enhancement. Due to the soft nature of the tumor and its elongation,25 the optic nerve is commonly kinked in the immediate retrobulbar zone (Figs 23.1, 23.3, and 23.5), a finding which helps to differentiate glioma from the rarer, but stiffer, extrinsic and generally more damaging childhood optic nerve sheath meningioma.26 Intraorbital optic nerve gliomas found in NF1 also often possess a characteristic feature: a superimposed reactive arachnoid proliferation tied to mucinous accumulation in the perineural subarachnoid space (Fig. 23.6).10,23 T2-weighted images of NF1 optic nerve gliomas thus much more commonly show an area of high signal intensity (corresponding to the mucinous/high water content element), the so-called “pseudo-CSF” sign, surrounding a central core of lower signal intensity (the intraneural tumor) (see Figs 23.1 and 23.3).25,27,28 Chiasmal gliomas are noted as an enlarged suprasellar mass with recognizable contours preserved, that may be accompanied by a diagnostic contiguous enlargement of the optic nerve or tract. The adjacent hypothalamus may also be involved.
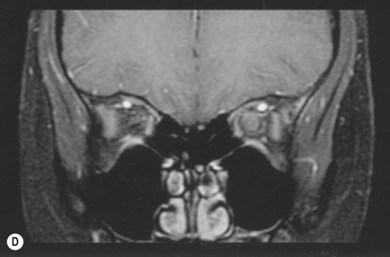
Fig. 23.5 Spontaneous regression of optic nerve glioma in the 14-year-old girl depicted in Figure 23.2. (A) MRI shows a fusiform enlargement of the left optic nerve, hyperintense and homogeneous in signal intensity from the globe up to the chiasm with intervening kink downward creating an apparent discontinuity. The left globe is proptotic and there is widening of the left neural foramen. (B) MRI reveals marked enlargement of the left intraorbital optic nerve, with prominent and homogeneous signal enhancement. (C) MRI 1 year later shows shrinkage of the optic nerve tumor along its entire course, with decrease in signal intensity. The left globe is no longer proptotic. (D) MRI shows clear reduction in optic nerve diameter, with only trace signal enhancement centrally. This girl had been scheduled to receive radiation therapy, but 1 week before this was to be initiated, she reported improved vision. Her planned radiation therapy sessions were cancelled while her vision improved from counting fingers at 4 feet, to 20/15 over the course of the year. Had the patient’s radiation therapy been scheduled a week earlier, her improvement would have erroneously been attributed to the treatment. (Patient of the Johns Hopkins Hospital). Anecdotal observations of tumor shrinkage noted following radiation or chemotherapy, particularly when noted long after termination of therapy, are likely due to the phenomenon of spontaneous regression.
Biological behavior and management
By definition, grade I pilocytic gliomas possess only rare, if any, mitotic figures and the majority of tumors demonstrate overall stability2,14,29 with only limited growth potential during development. Unlike higher grade gliomas, they do not show p53 mutations.30 They grow mainly by accumulation of mucosubstance, often with cystic enlargement and hydration.10,29 A few, however, may also show enlargement of the solid tumor component.31 On the other hand, spontaneous regression may also occur,11 and once a tumor is discovered, it is as likely to shrink given enough time, as it is to grow initially.11,32–36 Although as many as 25% of children with NF1 have gliomas,1,17 such incidences have not been noted in adults with NF1, and virtually no new cases present in adults with, or without, NF1. Misleading terminology is sometimes used to describe mostly adult and grade II and higher gliomas as “pilocytic-like,” “atypical pilocytic,” or “pilocytic with anaplastic features.”3,8 However, spontaneous anaplastic degeneration of grade I pilocytic astrocytomas does not occur. Reports purporting to show such changes have described only transformations iatrogenic in origin, secondary to the late effects of radiation.3 Gliomas also do not metastasize in the usual sense; during infancy, rarely, “drop metastases”, often asymptomatic in nature, may occur to the leptomeninges via the cerebrospinal fluid (CSF) passageways after surgical manipulations during ventricular shunt placement, or less commonly, after hemorrhagic cystic degeneration and rupture,37–40 much as occurs with such lesions noted intraocularly.41 Given these characteristics, these tumors fulfill the criteria for, and are best described as, glial hamartomas.2,3,42–45
Growth of gliomas is a function of both cellular proliferative and apoptotic activity; both may be high, or both low, reflecting a steady-state function within a tumor that demonstrates clinical stability.30,46 For these reasons, histopathologic examinations assessing proliferative activity alone,8,9,47,48 without also acertaining the rate of apoptosis, cannot provide information of prognostic utility.11,43 Inherently limited biopsy sampling sizes for tumors that are known to be heterogeneous both in their composition and in their growth patterns and phases, further limits the prognostic potential for such approaches.
Since mitoses are very rare, if at all present, no clinically significant benefit should be expected from antimitotic ionizing radiation beyond that expected from background spontaneous regression in the natural evolution of these tumors. Due to the severe adverse effects, moreover, of radiation on incompletely myelinated and still developing brain – including severe mental and growth retardation, psychiatric problems, vascular occlusions, and the induction of second tumors – attempts at treatment via ionizing radiation are now contraindicated in children.29,32,33,49,50 For similar reasons, antimitotic chemotherapy also offers no benefits. In previous studies reporting minor treatment effects, investigators failed to stratify results for those with “low-grade gliomas” into those with grade I pilocytic gliomas versus those with grade II fibrillary astrocytomas. As with initial reports ascribing benefits to radiation therapy, these studies also do not take into account the acknowledged fact of spontaneous regression11,42,51 and often include instances of tumor regression long after the cessation of therapy as a treatment effect.52 Loss of vision, moreover, is used in many treatment protocols as a measure to initiate chemotherapy. Declining acuity, however, does not correlate with tumor enlargement22,29 and can also result from the spontaneous regression of tumors distorting nerve axons. Subsequent immediate radiographic evidence demonstrating tumor size reduction then is erroneously attributed to an effect of treatment.22 More recent surveys and other studies, some of them specifically addressing in part some of these concerns, have confirmed the lack of beneficial treatment effects in the face of demonstrated toxicities.35,36,53
Optic nerve gliomas situated anterior to the chiasm may appear as threatening to involve this structure (see Fig. 23.5). Despite radiologic appearances, convincing evidence is lacking to show such evolution often occurs or that surgical excision of such tumors24 will prevent anticipated contralateral eye involvement; one may be witnessing instead multicentric nests of cells within different phases of growth (Fig. 23.1) rather than a true progression and invasion of cells moving forward.10
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


