Chapter 39 Uveitis
Introduction
This chapter will include endogenous childhood ocular inflammation including uveitis and the vasculitides. The differential diagnosis of childhood uveitis includes infection, hereditary anatomic abnormalities, and degenerations that may be accompanied by inflammation and tumors of childhood (Box 39.1). Ocular inflammation also accompanies systemic vasculitides; following infection (“reactive uveitis”); and accompanying congenital immunodeficiency syndromes and systemic autoinflammatory diseases.
Box 39.1
Differential diagnosis of childhood intraocular inflammation
Unreported trauma or intraocular foreign body
Photoreceptor dystrophy, especially where retinal pigmented epithelium (RPE) changes have not yet developed or where RPE changes resemble choroiditis
Hereditary vitreoretinal degeneration
Retinal vascular abnormalities that leak or bleed: Coats’ disease, vasoproliferative tumors
Congenital disk abnormalities especially with secondary vascular complications
Infection, especially in the congenitally or iatrogenically immunodeficient
Uveitis is a relatively common feature of several localized autoinflammatory diseases that have both unique and shared genetic associations (Table 39.1). There is often a family history of a wide range of autoimmune and autoinflammatory conditions in children with idiopathic uveitis unaccompanied by systemic disease. However, despite the wide variety of identified genetic associations, uveitis patients only rarely cluster in families, other than those with a high prevalence of HLA-B27, and there must be many environmental triggers and other genetic causes that remain unidentified.
General considerations
When considering ocular inflammation in childhood one needs to ask:
1. Why has the inflammation started in childhood rather than adulthood?
2. Will the age of onset influence the disease phenotype?
3. Is there a genetic disorder of the control of inflammation?
4. Is there a disorder of immunity predisposing to autoimmunity?
5. Is the inflammation secondary to a genetic degenerative process?
Familial clustering of unusual disease patterns suggests rare or novel mutations. The immune system develops through childhood and adolescence when most infections are first encountered. An unusual ocular inflammatory response to infection may be the first expression of a congenital systemic immune disorder (Box 39.2).
Organization of service
Childhood uveitis requires an organizational approach:
1. Early therapeutic decisions have to be made.
2. Effective management depends on primary care, pediatricians, and long-term social and educational planning.
3. Multiple surgical and medical specialties may be required.
4. Outcomes depend on public health measures as well as drugs and surgery.
5. General ophthalmologists need information about the threshold for tertiary referral. Late presentation and chronicity are common leading to high complication rates compared to similar conditions in adulthood.
6. In children, amblyopia, postsurgical glaucoma and fibrosis, the growth effects of parenteral steroids, and immunosuppressants pose special problems.
7. Compliance with treatment may be problematic for children and families.
Evaluation for systemic disease
Ocular inflammation may herald systemic disease (see Table 39.1). Physicians and ophthalmologists best work as teams. Symptoms of systemic disease may occur later than the ocular disease in oligoarticular juvenile idiopathic arthritis (JIA), sarcoidosis, Behcet’s disease, and Vogt-Koyanagi-Harada (VKH) syndrome.
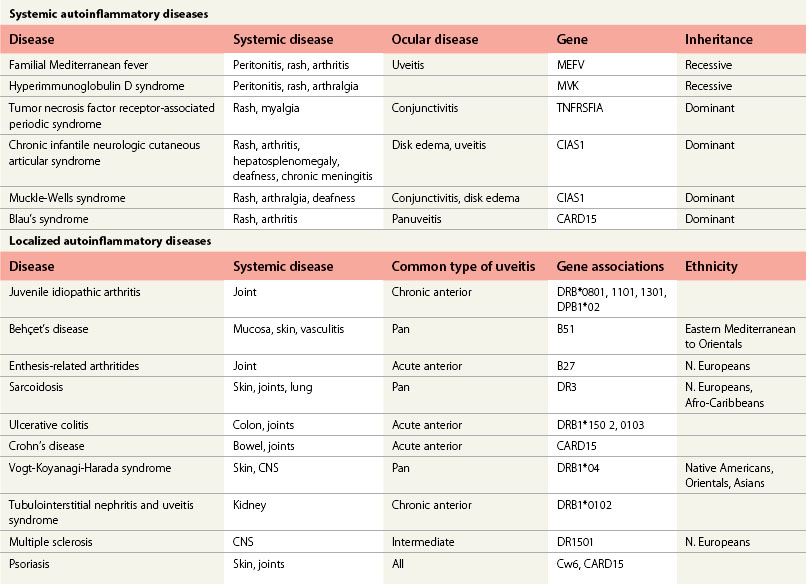
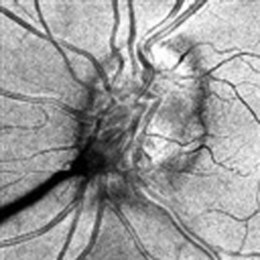
Associated central nervous system (CNS) inflammation is difficult to diagnose if it presents with behavioral changes, deafness, retrobulbar optic neuritis, headaches, or movement disorders in preverbal toddlers. Optic disk edema is frequent in childhood uveitis and scleritis and should not invariably require the exclusion of raised intracranial pressure (ICP) by lumbar puncture and neuroimaging. However, the diagnosis of optic disk changes (Figs 39. 1 and 39. 2) may be challenging and raised ICP can occur in pediatric sarcoidosis, systemic lupus erythematosus (SLE),1 chronic infantile neurologic cutaneous and arthritis (CINCA) syndrome, venous sinus thrombosis, and steroid use. Other causes of optic disk swelling include hypotony and optic neuritis.
Epidemiology of pediatric uveitis
Childhood uveitis is uncommon; rising with age. In 0- to 4-year-olds, the incidence is 3/100 000, in 10- to 14-year-olds 6/100 000, and in adults aged 17–25/100 000. It comprises 5% of most series of uveitis patients.2
The most common type of pediatric uveitis at a population level is idiopathic. The most common pattern in young children is chronic anterior uveitis; in those aged 8–16 it is intermediate uveitis. After age 16, the pattern of uveitis types is similar to that in adulthood (Box 39.3). Idiopathic chronic painful bilateral anterior uveitis, idiopathic anterior and intermediate uveitis, and idiopathic panuveitis are also not uncommon in children. Specific uveitis syndromes, such as birdshot retinochoroidopathy, are exceptionally rare in children.3
Box 39.3
Commonest types of uveitis by age
| 0–6 years | JIA-CAU |
| 7–12 years | Idiopathic CAU, IU |
| 13–16 years | IU, B27-associated AAU |
| Adult | B27-associated AAU |
Juvenile idiopathic arthritis (JIA) is the most common extraocular disease reported in tertiary referral series followed by those with no systemic disease (idiopathic uveitis), enthesitis related arthritis (ERA), sarcoidosis,4 inflammatory bowel disease (IBD), and Behçet’s disease. Even in countries with high rates of Behcet’s disease, Behcet’s uveitis is uncommon in childhood.5 VKH6 and tubulointerstitial nephritis and uveitis syndrome (TINU)7 can begin in childhood. Behçet’s disease and VKH syndrome are 10–100 times more common in some Oriental, Asian, and Mediterranean groups than in Caucasians.
Epidemiology of vasculitis
The commonest type of childhood systemic vasculitis is Henoch-Schonlein purpura which does not have ocular involvement;8 giant cell arteritis does not occur in childhood. The incidence of SLE is 0.8/10 000 and juvenile dermatomyositis 0.4/100 000. Childhood polyarteritis nodosa (PAN) is the fourth most common and ocular involvement is not uncommon. Wegener’s granulomatosis, Behçet’s disease, and microscopic polyangiitis are very rare – each less than 0.1/100 000.
Clinical types of uveitis
Idiopathic uveitis
Classification is best made on the basis of the site of visible inflammation: unilateral or bilateral, acute or chronic, painful or painless and redness present or not (Fig. 39.3). The anatomic classification and terms for clinical activity of uveitis have been revised.9 Patients with significant cell counts in both the anterior chamber and vitreous should be classified as “anterior and intermediate uveitis.” This describes some cases of JIA-uveitis and some previously classified as intermediate uveitis. Chronic unilateral uveitis suggests an infective cause but should not preclude a search for systemic disease. Twenty percent of JIA-uveitis remains unilateral.
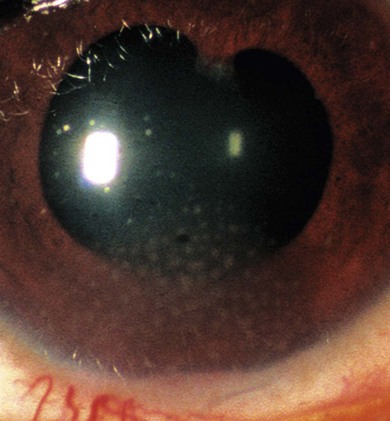
Painless anterior uveitis
Chronic painless AU, without systemic disease, is common in young children. The arthritis of JIA may develop up to 7 years after the onset of chronic painless AU (Fig. 39.4). Investigation of renal function, for TINU syndrome, sarcoidosis, and possible immunodeficiency is also indicated. Skin rashes are a common manifestation of sarcoid, Blau’s early onset sarcoidosis syndrome and CINCA: they are easier to biopsy. Fuchs’ heterochromic cyclitis is very rare in early childhood
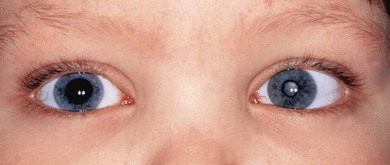
Intermediate uveitis
This refers to vitritis with a variable retinal inflammation and a minimal anterior segment inflammation (Table 39.2). The average age of onset is 9–13 years. Young children present late, and there may be extensive retinal complications rarely seen in adults. Children are less likely to develop macular edema but it is more difficult to resolve. Optic disk edema with chronic vitritis is more common than in adults. Peripheral vascular abnormalities are difficult to detect and may only be suspected when they cause hemorrhage. Neovascularization of the optic disk and retina may be caused solely by inflammation and subside with immunosuppression.10
Table 39.2 Structural complications of JIA-uveitis and IU
| JIA-uveitis | Intermediate uveitis |
|---|---|
| Anterior segment changes | |
| AC cells and flare | AC signs only in “anterior and intemediate uveitis,” and usually confined to cells only |
| Band keratopathy | |
| Extensive posterior synechiae | |
| Progressive anterior synechiae | |
| Iris hyperemia and rarely vascularization | |
| Unexplained hyphema | |
| Iris vascularized membrane | |
| Pupillary membrane | |
| Early irreversible ciliary body damage with chronic hypotony and phthisis | Hypotony rare |
| high risk glaucoma | Low risk glaucoma |
| Early cataract formation | Late developing despite persistent inflammation |
| Persistent diffuse vitreous flare and opacities with minimal vitritis | Vitritis necessary for diagnosis |
| Opacities tend to be dependent and aggregate – snowballs, snowbanks | |
| Diffuse macular and disk edema and subretinal fluid with minimal vitritis | Disk edema can be marked in absence of macular edema |
| Disk neovascularization | Disk and peripheral retinal neovascularization |
| Vitreous hemorrhage | Vitreous hemorrhage |
| Retinal detachment rare without | |
| Coexisting hypotony | Retinal detachment, pars plana cysts, localized peripheral detachments |
| Retinoschisis | |
| Postoperative complications | |
| IOL membrane formation | Usually tolerates IOL |
| Universal posterior capsular opacification | |
| Membrane formation following capsulotomy |
Panuveitis and multifocal choroiditis
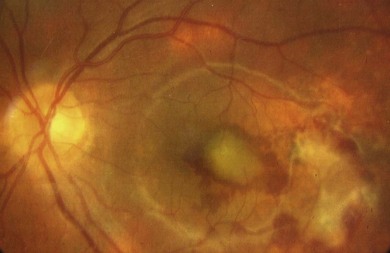
Unifocal, unilateral choroiditis suggests infection such as toxoplasmosis. Multifocal, bilateral disease suggests systemic disease or a white spot syndrome, both rare in childhood. Many children with multifocal choroiditis and vitritis resemble sarcoidosis, with no evidence of extraocular disease. Some infections such as brucellosis, borreliosis, and varicella may also mimic sarcoid choroiditis. Multifocal choroiditis and panuveitis rarely occurs in childhood. It can be complicated by severe visual loss from secondary choroidal neovascularization and warrants aggressive control of inflammation (Fig. 39.5). If the fundus cannot be seen in children with AU it must be assumed they have panuveitis. Ultrasound may disclose scleritis or focal infection or tumor.
Retinitis
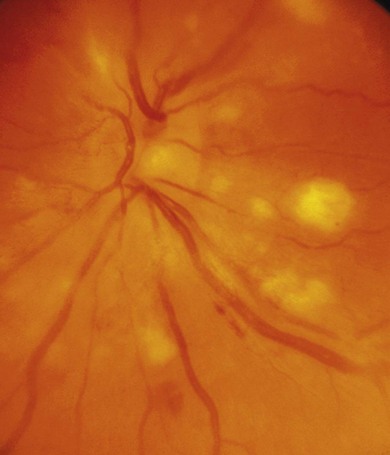
Focal infiltrates of the inner retina with little overlying vitritis are typical of acute viral infections (Fig. 39.6). They are more discrete and persistent and yellower than cotton-wool spots. Retinal infiltrates in Behçet’s disease always have signs of retinal vasculitis.
Neuroretinitis
In neuroretinitis, swelling of the neuroretina, maximal at the optic disk, is out of proportion to signs of posterior segment inflammation (see Chapter 53). Exudates around the fovea form a macular star. Children are more likely to develop edema in response to infections that trigger optic neuritis and neuroretinitis. Cat-scratch disease typically involves the posterior segment in this manner.
Parainfectious uveitis
Childhood is a time when airborne pathogens are first encountered and transient ocular inflammation may result. Varicella and streptococcal infections are frequent causes of reactive uveitis. Reported associations may be coincidental and identification of intraocular viruses in specific uveitis types remains rare. Clusters of severe acute panuveitis in children have been reported from Tanzania and Nepal; in the former the incidence was 540/100 000 in the under 9s.5–8
Acute anterior uveitis (AAU) is the most common ocular inflammation following varicella but mild retinal vasculitis, a self-limiting retinitis, and multifocal choroiditis can occur, usually within 4 weeks (Fig. 39.6). Chronic inflammation and progressive retinitis is an absolute indication for antiviral treatment, not usually necessary for AAU.
Localized autoinflammatory diseases
Juvenile idiopathic arthritis
Epidemiology
The incidence of juvenile arthritis is 10/100 000. Only half are patients with oligoarticular or polyarticular JIA, and the incidence of JIA-uveitis is 1/100 000 (Table 39.2).
JIA types associated with CAU
Genetic studies suggest similarities between oligoarticular and polyarticular JIA in the youngest groups.11 Older children with ANA-negative polyarticular JIA may have a distinct pattern of disease and appear to be at a much lower risk of CAU. JIA is found in all races; JIA-uveitis may be more common in Caucasians.
Several genes, including HLA, are associated with different clinical types of JIA.13,14 Oligoarticular onset JIA is associated with HLA genes common to polyarticular JIA (DRB1*08), psoriatic arthropathy (DRB1*1301), and systemic onset JIA (DRB1*11), as well as unique associations such as DPB1*02. Uveitis appears to be associated primarily with the DRB1*13 haplotype, which is most frequent in oligoarticular JIA, and DPB1*02. ERA and early onset rheumatoid arthritis are clinically and genetically distinct from the types of JIA associated with CAU.
In those developing uveitis, arthritis typically starts at 28 months and uveitis 13 months later: 86% have oligoarticular onset JIA, 75% are female, and 80% are ANA-positive.10,12



