Chapter 131 Tuberous Sclerosis and the Eye
Introduction
History, diagnosis, and genetic basis
Tuberous sclerosis complex (TSC) is a rare multisystem genetic disorder, characterized by hamartomatous tumors of the brain, skin, viscera, and eye. It is inherited in an autosomal dominant fashion,1 with a high degree of penetrance2 with variable phenotypic expression. The first publication of a color plate by Rayner of a patient with an apparent facial angiofibroma took place in 1835.3 In 1880, D.M. Bourneville, from a neuropathological study of a young patient with seizures, hemiplegia, mental subnormality, who also had renal tumors, coined the term tuberous sclerosis.4 However, the eponym Bourneville’s disease was commonly used to describe this condition until only recently. In 1908, Vogt5 first proposed epilepsy, mental retardation, and the skin lesions of adenoma sebaceum as a diagnostic triad. In 1920, Van der Hoeve6 first recognized retinal involvement in tuberous sclerosis. Ophthalmologists now recognize astrocytic hamartomas of both the retina and the optic nerve as common features of this condition.
The diagnostic criteria are based on the premise now that there are no truly pathognomonic features or clinical criteria for the diagnosis of tuberous sclerosis or tuberous sclerosis complex (TSC).5 Signs that were once regarded as sufficiently specific are now known to occur as isolated findings in normal individuals without TSC.7 In theory, the perfect disease classification scheme should have both a high sensitivity (low rate of missing the diagnosis of tuberous sclerosis) and a high specificity (low rate of incorrectly labeling a patient with tuberous sclerosis). The committee recommended the adoption of an ordinal classification of tuberous sclerosis: definite, probable, and suspect tuberous sclerosis.7 Patients can be classified into one of three categories based on the presence of clinical features of varying specificity (primary features: high specificity; secondary features: moderate specificity; tertiary features: lower degree of specificity).8 The revised criteria require either TSC-associated lesions in two or more organ systems or two dissimilar lesions in one organ system to confirm the diagnosis. The classic triad included seizures, mental retardation, and cutaneous angiofibromas, which, however, only occurs in 29% of cases.9 Mental retardation and seizures were also left out of the revised criteria by the 1998 expert consensus panel.10 The addition of DNA testing complements the clinical diagnosis and helps with genetic counseling, especially in concert with prenatal testing.11
Systemic manifestations
Neurological
Seizures
Children with tuberous sclerosis often have seizures that are referred to as “infantile spasms” or “salaam attacks.”7 These seizures are characterized by repetitive myoclonic spasms of the head, neck, and limbs. Originally described by West,12 an English GP, through the experience of his own son in 1841, the seizures last for seconds but may occur in bouts of 10–50. Hoyt13 described infantile spasms as “lightning fast nodding of the head, often with extension or flexion of the trunk and frequently also of the arms.” According to Pampliglione and Pugh,14 25% of children affected by “infantile spasms” develop other stigmata of tuberous sclerosis within 4 years of diagnosis. Infantile seizures, the most common feature of the disease, tend to evolve into grand mal seizures at a later time and are seen in 93% of patients.11,15 Presently, the treatment of epilepsy in TSC remains a major challenge, even with new anticonvulsant medications.16 Even after neurosurgery, seizures recur in approximately 30% of cases.17
Cognitive and behavioral disability
Children with tuberous sclerosis may have normal or above-average intelligence.15 However, Lagos and Gomez15 reported that 44 (62%) of 71 tuberous sclerosis patients were labeled as mentally retarded. Subsequent reports confirm that neurocognitive manifestations ranging from profound cognitive disabilities to mild learning impairment occur in around 45% of patients.18 While the majority of individuals with TSC have normal intelligence, they may still be prone to specific cognitive memory, attentional and executive skills deficits.19 It should be noted that early reports emphasizing the prevalence of mental retardation in tuberous sclerosis suffered from selection bias because they were based on patients who were institutionalized.11 In the Lagos and Gomez series,15 intracranial calcifications were more commonly seen in patients with normal intelligence. The cause of both seizures and mental retardation may be related to the presence of cortical tubers (Fig. 131.1) and subependymal hamartomas (Fig. 131.2). These tumor masses represent benign astrocytic hamartomas, which characteristically involve the basal ganglia, lateral and third ventricles, and the posterior fossa. Histopathologically, they consist of well-differentiated large astrocytes in an admixture of fibrillary astrocytes and calcospherules.20 They frequently undergo cystic degeneration and dystrophic calcification, accounting for their typical radiologic features (Fig. 131.3) and for the name tuberous sclerosis.21 Most significant variables associated with a poor cognitive outcome include a history of refractory seizures, TSC2 mutations, and cortical tubers in specific locations of the brain.22
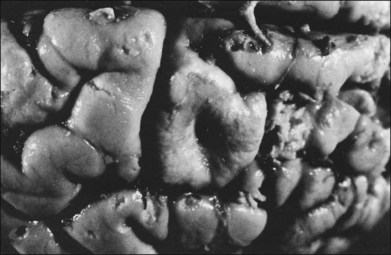
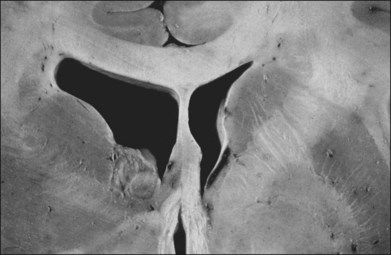
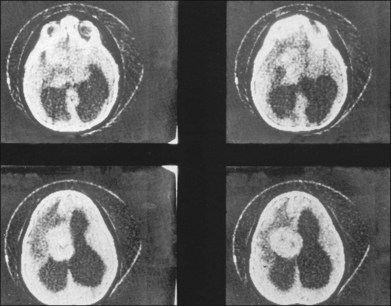
Brain tumors are associated with tuberous sclerosis. These well-circumscribed lesions are classified as subependymal giant-cell astrocytomas and should not be confused with the giant-cell variant of glioblastoma multiforme, a malignant astrocytoma.23
A case of giant cerebral aneurysm with visual loss as a result of optic nerve compression in a patient with tuberous sclerosis has been reported.24
Skin features
Facial angiofibromas formerly referred to as adenoma sebaceum, occur as a reddish-brown papular rash found characteristically in a “butterfly” distribution over the face. This rash is a pathognomonic hallmark of tuberous sclerosis and is very sensitive, occurring in over 85% of patients.15 Histopathologically, adenoma sebaceum consists of multiple smooth papules that are benign angiofibromas.25 The rash is generally not apparent at birth but appears in childhood, generally before the age of 9 years, and tends to intensify with time.20 It is most prominent in the nasolabial folds and over the malar areas.
The skin lesion formerly believed to be pathognomonic for tuberous sclerosis is the hypopigmented macule or patch (Fig. 131.4). These lesions are variable in shape, with only an occasional one justifying the term ash-leaf sign. Often present at birth, this is the first clinical sign of disease. Its occurrence is highly variable, being present in up to 75% of affected children.26 Pathologically, the ash-leaf lesion is an achromic nevus,27 as opposed to vitiligo, in which the melanocytes are actually missing. Ultraviolet light (classically a Wood’s lamp) may be used effectively in a darkened room to screen for the ash-leaf sign.28 The importance of this clinical sign in the workup of a child with infantile spasms of unknown cause cannot be overemphasized.26
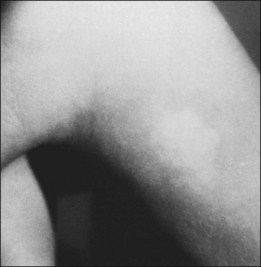
Fig. 131.4 Hypopigmented skin macule or patch (achromic nevus), sometimes referred to as an ash-leaf sign.
(Courtesy of P. McLeod.)
Shagreen patches represent areas of skin affected by fibromatous infiltration. This sign is seen in approximately 20% of cases, often in the lower back.29 This flesh-colored, leathery plaque on the lumbosacral area is highly characteristic of the tuberous sclerosis complex.28 Subungual fibromas may be seen on the hands and feet.15,28
Visceral features
Although Vogt’s classic triad of mental deficiency, epilepsy, and facial angiofibromas implies central nervous system and skin involvement, it is important to be cognizant of the possible presence of visceral tumors. Renal angiolipomas are hamartomas that occur singularly or in multiples, in unilateral or bilateral fashion. They are present in 80% of patients with tuberous sclerosis.20 They are benign tumors that are not known to metastasize to distant sites,30 although extension to the renal vein has been noted.31 Cardiac hamartomas also occur. They are rhabdomyomas that are single or multiple whitish nodules that characteristically protrude into the ventricles.20 Slowly, progressive subpleural cysts may form and can rupture, causing a spontaneous pneumothorax.32 Hamartomas of the liver, thyroid, pancreas, and testis have been reported.20
Skeletal features
The skeletal system is affected in 40% of cases.7 Sclerotic calcified areas are seen in the skull and spine. Skull radiographs may show opacities over the calvaria that represent intraosseous sclerotic areas of intracranial calcified astrocytic hamartomas. Computed tomography (CT) scans are particularly helpful in the investigation of tuberous sclerosis.33 Phalangeal cysts affect the hands and feet.7
Ocular manifestations
Retinal manifestations
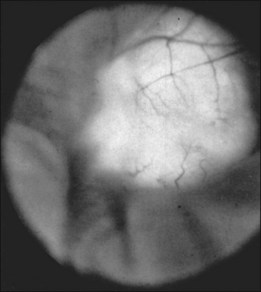
The retinal phakomas of tuberous sclerosis are astrocytic hamartomas of the retina, seen in an estimated 53% of patients, according to Lagos and Gomez15 in their Mayo Clinic series of 71 cases. Shelton34 found evidence of retinal hamartomatous lesions in all seven patients examined. Solitary astrocytic hamartomas may be seen in otherwise healthy patients, but in patients with tuberous sclerosis they may be multifocal or bilateral. Although most frequently seen in the setting of tuberous sclerosis, these hamartomas may also be noted in patients with neurofibromatosis.35,36 Although astrocytic hamartomas of the retina are the principal ocular manifestation of the tuberous sclerosis complex, two patients have been reported in whom stromal depigmentation of the iris and atypical colobomata were correlated on histopathological examination with hamartomas of the iris pigment epithelium and ciliary body epithelium.37
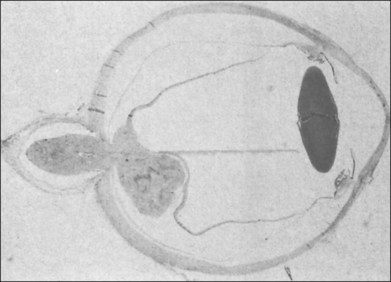
Typically, astrocytic hamartomas can be classified morphologically as one of two types: (1) large, whitish (calcified) nodular masses or (2) flat, translucent (noncalcified) smooth tumors.38 Nyboer et al.39 have described an intermediate type of retinal hamartoma having features of both types. These lesions typically occur at or near the optic disc. More peripheral lesions may occur, and these may be more easily confused with retinoblastoma. Although almost all astrocytic hamartomas of the retina in tuberous sclerosis are endophytic in nature, an exophytic case has been described (Figs 131.5, 131.6 illustrate a different case).31 This case consisted of the pathologic examination of an eye enucleated for neovascular glaucoma from a 10-year-old boy with tuberous sclerosis and seizures. The retina was totally detached and was associated with a large multinodular mulberry-like astrocytic tumor in the subretinal space.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


