Chapter 38 The uveal tract
Symptoms of uveal disease
Congenital structural and developmental abnormalities of the pupil include iridodonesis, aniridia, micro pupil, polycoria, corectopia, ectopia, coloboma, iris sphincter atrophy, and dilated oval pupils (Box 38.1, Figs 38.1 and 38.2). Acquired abnormalities include trauma (Fig. 38.3), ischemia, hemorrhage, or involvement with tumors such as lymphoma, leukemia, juvenile xanthogranuloma, leiomyoma, and neurofibromas.
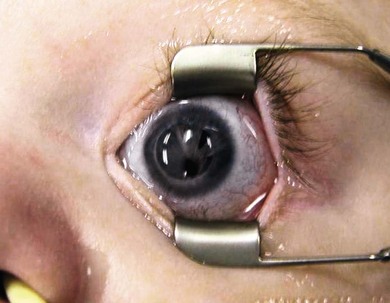
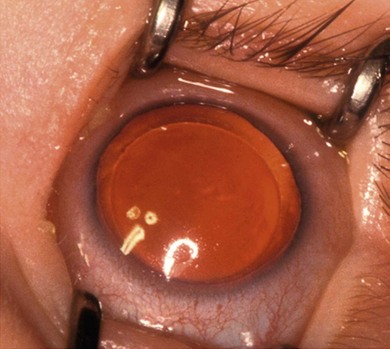
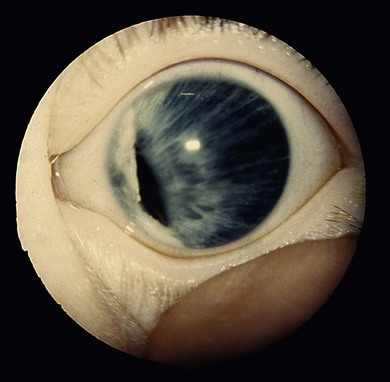
Anisocoria is a difference in the size of the pupils. It occurs in up to 20% of the normal population. It may be transmitted as an autosomal dominant trait with variable expression.1
Congenital idiopathic microcoria, an abnormality of development of the pupillary membrane, is usually unilateral with an almost absent eccentric pupil. It may be an autosomal dominant trait. It is associated with myopia and corectopia.2 Deformed pupils may be caused by unequal growth of neural crest cells in parts of the iris stroma. Bilateral microcoria has been reported in association with microphthalmos and posterior anomalies.3 Early surgical treatment and occlusion therapy can result in useful vision.4
Persistent pupillary membranes
Congenital persistent pupillary membranes result from incomplete involution of the tunica vasculosa lentis which does not involute until the third trimester and is seen in premature babies. They are mostly sporadic. They are formed by buds from the annular iris vessels growing centrally to form the anterior vascular tunic of the lens.5 They may span the entire pupil and be attached to the anterior capsule of the lens with or without a cataract. They may occur with microcornea, megalocornea, microphthalmos, and coloboma. Persistence of extensive membranes is uncommon. Most are asymptomatic and require only a mydriatic or no treatment.6 However, it is sometimes difficult to assess whether a membrane is amblyogenic. Removal involves surgery with viscoelastics and excision with microscissors7 or a vitrector. A cataract may be induced if the lens is damaged, and the surgeon must avoid cutting the iris or creating an iridodialysis.8,9 Laser lysis uses a Nd:YAG laser to disrupt the adhesions between the strands of the membrane and the normal iris.10 Pigment dispersion and hyphema are potential complications. The visual prognosis can be good,11 but poor initial visual acuity is an indication of a poor outcome. Myopic and hyperopic anisometropia is more prevalent in eyes with more severe hyperplastic persistent pupillary membranes. A 5-week-old baby presented with persistent pupillary membranes in her left eye (Fig. 38.4). They seemed amenable to medical treatment so we used pupil dilation and patching. At 4 months, she was intolerant of patching. We removed the membranes through a small incision using viscoelastics and capsulotomy scissors. She had a clear visual axis postoperatively and tolerated patching. She now has equal vision in both eyes. A 10-year-old child presented with pupillary membranes in both eyes (Fig. 38.5). He was photophobic and had reduced vision. His pupils dilated poorly and it was impossible to see his fundi or assess his refraction. Surgical removal was performed using viscoelastics and a capsulotomy scissors. Four years later he has a corrected vision of logMAR 0.3 (6/12, 20/40, 0.50) and is moderately myopic.
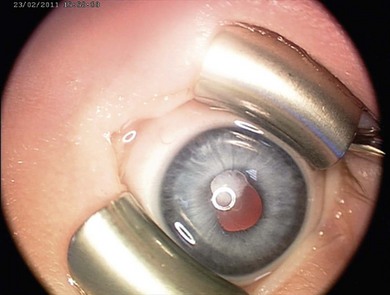
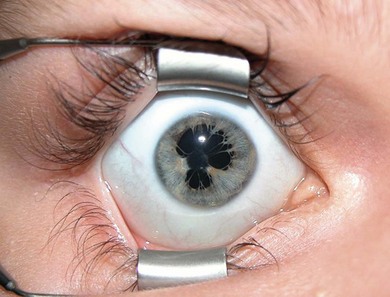
Congenital iris and stromal cysts
Iris cysts may be congenital resulting from an accumulation of fluid in an epithelium lined cyst.12 They can be in the pigment epithelium or stroma and have a squamous epithelial or neuroepithelial lining.13 Iris pigment cysts are usually stationary and have a transparent wall with a vascular lining and occur at the pupillary margin. Stromal cysts are progressive, in the anterior surface of the iris. Whilst some advocate a conservative approach, many require surgery.13 The main treatment options are aspiration with or without cryotherapy applied to the base of the cysts near the limbus, injection of sclerosant (risky), surgical excision with sector iridectomy, Nd:YAG or argon laser, or irradiation.14,15 The differential diagnosis of congenital iris cysts includes cysts from epithelial implantation due to surgery, trauma, and iris or ciliary body tumors.16 Complications include glaucoma,17 cataract,18 and spontaneous detachment to become free-floating.19 Ciliary body cysts are less common; they may be solid or filled with fluid and may cause astigmatism and amblyopia. A neonate presented with a large iris stromal cyst encroaching on the visual axis (Fig. 38.6). At 6 weeks it was drained and partially excised. Three years later it remained small and he has excellent vision in the eye.
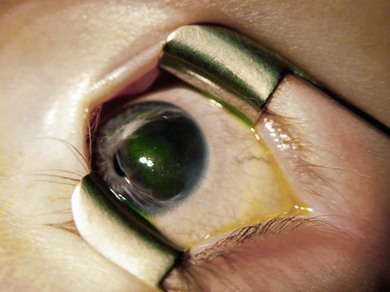
Iris ectropion or ectropion uveae
This is when the posterior pigment epithelium of the iris extends to the front of the iris. It may be congenital or acquired.20 It is sometimes associated with glaucoma, neurofibromatosis type 1 (NF1), or anterior segment dysgenesis. Iris flocculi are excrescences of the pigment epithelium at the pupil margin. They may be a marker of familial aortic dissection.21 A 2-year-old girl was referred with raised intraocular pressure (IOP) and a grossly cupped disk. She had ectropion uveae. She had a mitomycin-enhanced trabeculectomy which controlled her IOP to 8 mm, bleb revision 1 year later, and her vision improved to logMAR 0.48 (6/18, 20/60, 0.33) equivalent with Sheridan Gardner (Fig. 38.7).
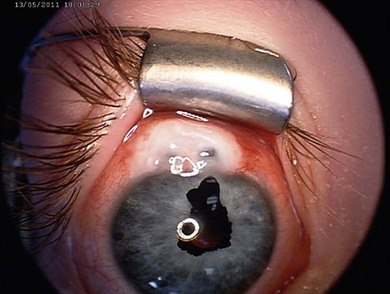
Heterochromia iridis
A difference in iris color can be congenital or acquired (Fig. 38.8). In congenital heterochromia the involved iris is darker, an indicator of ocular melanocytosis, oculodermal melanocytosis, or a sector hamartoma syndrome. Congenital Horner’s syndrome (see Chapter 63) results in ipsilateral hypopigmentation, miosis, and ptosis. Waardenberg’s syndrome (WS) (Fig. 38.9) is an autosomal dominant condition with sensorineural deafness, white forelock, and heterochromia iridis. Type 1 WS includes lateral displacement of the inner canthi, prominent root of the nose, and unusual brows. It is caused by a mutation in the PAXax 3 gene.22 Type 2 WS does not include facial dysmorphism: the gene has been located to 3p12-p141.23 Other types are rarer.
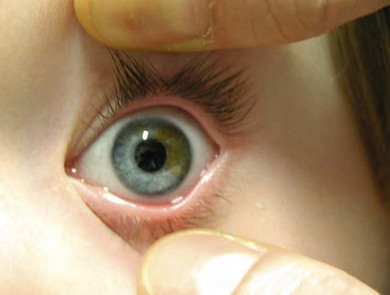
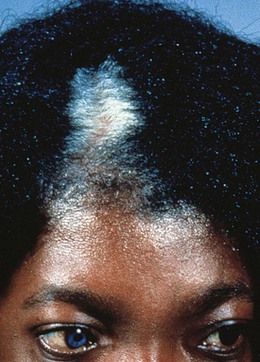
Iris sector heterochromia and Hirschsprung’s disease is an autosomal recessive condition representing a neural crest cell lineage abnormality.24,25
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


