Fig. 12.1
Anatomy of the bony orbit: (a) lateral wall, (b) floor, (c) roof, and (d) medial wall. FB frontal bone, FZF frontozygomatic fissure, gSB greater sphenoid bone, ZB zygomatic bone, IOF inferior orbital fissure, LF lacrimal fossa, MB maxillary bone, sof superior orbital foramen, NB nasal bone, EB ethmoidal bone, LB lacrimal bone, iof inferior orbital foramen; horizontal black arrow: posterior lacrimal crest; vertical black arrow, anterior lacrimal crest; horizontal white arrow, anterior ethmoidal foramen; vertical white arrow, posterior ethmoidal foramen; black asterisk, superior orbital notch; white asterisk, optic canal (Reproduced from: Karcioglu [460])
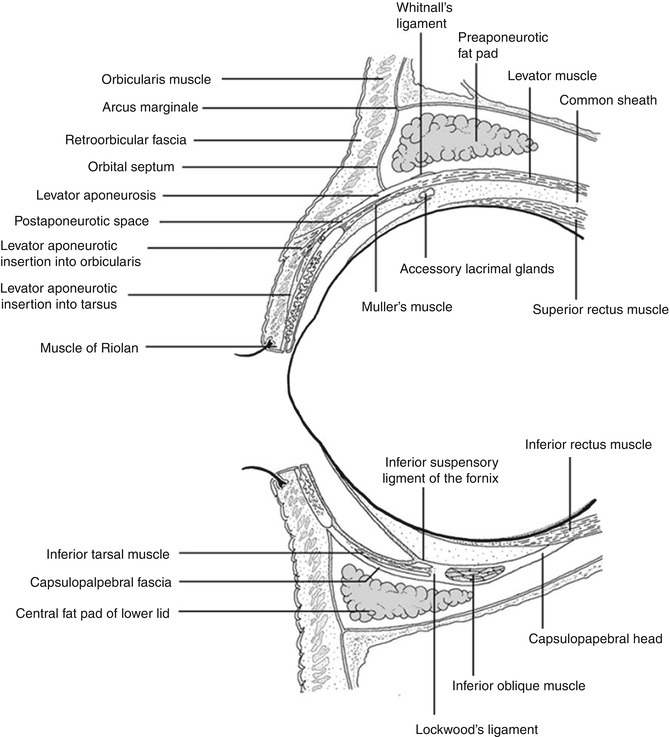
Fig. 12.2
Anatomy of the orbital cavity: central sagittal drawing of the orbital cavity including the orbital septum and the eyelids (Reproduced from Naumann et al. [461])
12.1.1 Classification and Frequency
The nucleus of this chapter is a consecutive series of 1,077 orbital biopsies and resections retrieved from the files of the Department of Pathology of the Erasmus MC University (the Netherlands) for the 25-year period, 1987–2012. This series proved a logical source for illustrations to aid the ophthalmic pathologist in daily diagnostic practice. Other illustrations were obtained from the Department of Experimental Medicine, University of Rome “Sapienza,” Rome, Italy, and cases presented at the European Ophthalmic Pathology Society and Verhoeff-Zimmerman Society or other ophthalmic pathology meetings as referenced. The most frequent lesions of the orbit in our series are of an inflammatory nature (29 %), closely followed by cysts (13 %), epithelial lesions (14 % primary and secondary), and lymphomas (10 %) (Table 12.1). Lesions of the soft tissue and bone lead to the most diverse number of diagnoses, of which vascular, meningeal, and neurogenic tumors are the most frequent. Metastasis represents 3 % of all orbital tumors. We have considered all orbital lesions including those of the lacrimal gland (N = 216, Table 12.2). The lesions of the lacrimal sac were evaluated separately (N = 238, Table 12.3).
Table 12.1
Histopathologic diagnoses of orbital lesions (total N = 1,077)
Diagnosis (category) | N | Relative % | Absolute % |
|---|---|---|---|
Cysts | 139 | 100 | 13 |
Dermoid | 92 | 66 | 8.5 |
Epidermoid | 9 | 6.5 | <1 |
Inclusion | 2 | 16 | 2 |
Eccrine | 1 | <1 | <1 |
Respiratory | 1 | <1 | <1 |
Pseudocyst | 1 | <1 | <1 |
Lacrimal gland | 24 | 17 | 1.6 |
Dacryops | 3 | 2 | <1 |
Nasolacrimal duct | 2 | 1 | <1 |
Mucocele | 4 | 3 | <1 |
Degenerations | 69 | 100 | 6.5 |
Fat prolapse | 16 | 23 | 1.4 |
Lacrimal gland prolapse | 8 | 12 | <1 |
Scar/fibrosis | 19 | 28 | 1.7 |
Cholesterol granuloma | 10 | 14 | 1 |
Hematic cyst | 4 | 6 | <1 |
Necrosis | 4 | 6 | <1 |
Radiation change | 2 | 3 | <1 |
Amyloid | 2 | 3 | <1 |
Xanthoma | 1 | 1 | <1 |
Oncocytic metaplasia | 1 | 1 | <1 |
Thromboembolism | 1 | 1 | <1 |
Dacryolith | 1 | 1 | <1 |
Inflammation | 310 | 100 | 29 |
Infection | 10 | 3 | <1 |
Abscess | 5 | 1.5 | <1 |
Cellulitis | 1 | <1 | <1 |
Osteomyelitis | 1 | <1 | <1 |
Chronic inflammation ECI | 40 | 13 | 1 |
IOI | 92 | 29.5 | 8.5 |
Florid lymphoid hyperplasia | 23 | 7 | 2 |
Sarcoidosis | 29 | 9 | 2.5 |
Xanthogranuloma | 17 | 5 | 1.5 |
Wegener’s granulomatosis | 12 | 4 | 1 |
Sjögren syndrome | 4 | 1 | <1 |
Churg–Strauss | 1 | <1 | <1 |
Giant cell vasculitis | 1 | <1 | <1 |
Pyogenic granuloma | 4 | 1 | <1 |
Granulation tissue | 12 | 4 | 1 |
Granuloma NOS | 1 | <1 | <1 |
Foreign body granuloma | 13 | 4 | 1 |
Rheumatoid arthritis granuloma | 1 | <1 | <1 |
Graves’ orbitopathy | 5 | 1.5 | <1 |
Paraffinoma | 8 | 2.5 | <1 |
Florid subperiosteal reaction | 1 | <1 | <1 |
Dacryoadenitis | 19 | 6 | 2 |
Dacryocystitis | 10 | 3 | <1 |
Muscle | 5 | 100 | 1.5 |
CPEO | 3 | 60 | <1 |
Hypertrophy | 1 | 20 | <1 |
Atrophy | 1 | 20 | <1 |
No diagnosis/normal | 37 | – | 3 |
Vascular | 66 | 100 | 6 |
Cavernous hemangioma | 37 | 56 | 3 |
Capillary hemangioma | 7 | 11 | <1 |
AVM | 6 | 10 | <1 |
Varix | 6 | 10 | <1 |
Lymphangioma | 5 | 7.5 | <1 |
Angiofibroma | 2 | 3 | <1 |
Epithelioid hemangioma | 1 | 1.5 | <1 |
Angiomyxomas | 1 | 1.5 | <1 |
Masson tumor | 1 | 1.5 | <1 |
Meningioma | 58 | 100 | 5 |
Meningothelial | 26 | 45 | 2 |
Transitional | 19 | 33 | 2 |
Psammomatous | 4 | 7 | <1 |
Fibrous | 2 | 3.5 | <1 |
Microcystic | 2 | 3.5 | <1 |
Chordoid | 3 | 5 | <1 |
Anaplastic | 2 | 3.5 | <1 |
Neurogenic | 27 | 100 | 2.5 |
Neurofibroma | 12 | 44 | 1 |
Traumatic neuroma | 5 | 19 | <1 |
Schwannoma | 3 | 11 | <1 |
Granular cell tumor | 1 | 1.5 | <1 |
MPNST | 1 | 1.5 | <1 |
Ganglioneuroma | 1 | 1.5 | <1 |
Optic glioma | 1 | 1.5 | <1 |
Olfactory neuroblastoma | 1 | 1.5 | <1 |
Retinoblastoma | 1 | 1.5 | <1 |
Glioblastoma | 1 | 1.5 | <1 |
Bone and soft tissue | 58 | 100 | 5 |
Chondroma | 1 | 1.5 | <1 |
Chondrosarcoma | 7 | 12 | <1 |
Osteoma | 6 | 10 | <1 |
Osteoid osteoma | 1 | 1.5 | <1 |
Osteosarcoma | 1 | 1.5 | <1 |
Osteopetrosis | 1 | 1.5 | <1 |
Ossifying fibroma | 1 | 1.5 | <1 |
Fibrous dysplasia | 7 | 12 | 1 |
Nodular fasciitis | 1 | 1.5 | <1 |
Fibromatosis | 2 | 3 | <1 |
IMT | 1 | 1.5 | <1 |
Haemangiopericytoma | 3 | 5 | <1 |
Fibrous histiocytoma | 3 | 5 | <1 |
Malignant fibrous histiocytoma | 1 | 1.5 | <1 |
Dermolipoma | 4 | 7 | <1 |
Spindle cell lipoma | 1 | 1.5 | <1 |
Liposarcoma | 2 | 3 | <1 |
Leiomyoma | 1 | 1.5 | <1 |
Leiomyosarcoma | 1 | 1.5 | <1 |
Embryonal rhabdomyosarcoma | 7 | 12 | <1 |
Alveolar rhabdomyosarcoma | 1 | 1.5 | <1 |
Glomus tumor | 2 | 3 | <1 |
Sarcoma NOS | 1 | 1.5 | <1 |
Ewing sarcoma | 1 | 1.5 | <1 |
Myositis ossificans | 1 | 1.5 | <1 |
Melanoma | 16 | 100 | 1.5 |
Conjunctival | 5 | 31 | <1 |
Skin | 4 | 25 | <1 |
Uveal | 6 | 38 | <1 |
Nose | 1 | 1 | <1 |
Hematopoietic | 105 | 100 | 10 |
Malt/marginal zone | 43 | 41 | 4 |
Follicular | 22 | 21 | 2 |
DLBCL | 13 | 12 | 1 |
Mantle cell | 9 | 8.5 | 1 |
Lymphoplasmacytic | 4 | 4 | <1 |
B-CLL | 3 | 3 | <1 |
Plasmacytoma | 3 | 3 | <1 |
Burkitt | 1 | 1 | <1 |
T cell | 1 | 1 | <1 |
LCH | 4 | 4 | <1 |
Chloroma/AML | 2 | 2 | <1 |
Epithelial (primary/secondary) | 149 | 100 | 14 |
Apocrine hidradenoma | 1 | 0.5 | <1 |
Oncocytoma | 1 | 0.5 | <1 |
Inverted papilloma | 10 | 7 | 1 |
Undifferentiated carcinoma | 32 | 21.5 | 3 |
Adenocarcinoma NOS | 5 | 3 | <1 |
Basal cell carcinoma | 11 | 7 | <1 |
Squamous cell carcinoma | 37 | 25 | 3.5 |
Basosquamous carcinoma | 1 | 1 | <1 |
Adenosquamous | 1 | 1 | <1 |
Verrucous | 1 | 1 | <1 |
Sebaceous | 10 | 7 | 1 |
Pleomorphic adenoma | 23 | 15 | 2 |
AdenoCa ex PA | 2 | 1 | <1 |
Squamous Ca ex P.A. | 1 | 0.5 | <1 |
Adenoid cystic carcinoma | 1 | 0.5 | <1 |
Acinic cell carcinoma | 1 | 0.5 | <1 |
Polymorphous low-grade adenocarcinoma | 1 | 0.5 | <1 |
Sinonasal adenocarcinoma | 5 | 3 | <1 |
Sweat gland carcinoma | 2 | 1 | <1 |
Merkel cell carcinoma | 1 | 0.5 | <1 |
Small cell carcinoma | 1 | 0.5 | <1 |
Transitional cell carcinoma | 1 | 0.5 | <1 |
Metastasis | 36 | 100 | 3 |
Mammacarcinoma | 13 | 36 | 1 |
Neuroendocrine carcinoma | 5 | 14 | <1 |
Undifferentiated carcinoma | 3 | 8 | <1 |
Adenocarcinoma NOS | 3 | 8 | <1 |
Thyroid carcinoma | 3 | 8 | <1 |
Melanoma skin | 3 | 8 | <1 |
Prostate carcinoma | 2 | 6 | <1 |
Salivary gland adenocarcinoma | 1 | 3 | <1 |
Pituitary carcinoma | 1 | 3 | <1 |
Lung carcinoma | 1 | 3 | <1 |
Nasal cavity carcinoma | 1 | 3 | <1 |
Miscellaneous | 2 | – | <1 |
Teratoma | 2 | – | <1 |
Table 12.2
Histopathologic diagnoses of lacrimal gland lesions (N = 216)
Diagnosis (category) | N | Relative % | Absolute % |
|---|---|---|---|
Cysts | 31 | 100 | 14 |
Lacrimal gland cyst | 24 | 77 | |
Dacryops | 3 | 10 | |
Dermoid cyst | 2 | 6.5 | |
Epidermoid cyst | 1 | 3 | |
Squamous cell carcinoma | 1 | 3 | |
Degenerations | 5 | 100 | 2 |
Fibrosis | 2 | 40 | |
Dacryolith | 1 | 20 | |
Oncocytic metaplasia | 1 | 20 | |
Thromboemboli | 1 | 20 | |
Inflammation | 106 | 100 | 49 |
Dacryoadenitis | 44 | 42 | |
IOI, sclerosing dacryoadenitis | 29 | 27 | |
Sarcoidosis | 16 | 15 | |
Lymphoid hyperplasia | 9 | 8.5 | |
Wegener’s granulomatosis | 3 | 3 | |
Sjögren syndrome | 2 | 2 | |
Xanthogranuloma | 1 | 1 | |
Rheumatoid granuloma | 1 | 1 | |
Giant cell vasculitis | 1 | 1 | |
No diagnosis | 17 | – | 8 |
Lymphoma | 26 | 100 | 12 |
Malt | 12 | 46 | |
Follicular | 5 | 20 | |
Mantle cell | 5 | 20 | |
DLBCL | 3 | 12 | |
Lymphoplasmacytic | 1 | 4 | |
Epithelial | 30 | 100 | 14 |
Pleomorphic adenoma | 16 | 53 | |
AdenoCa ex PA | 2 | 7 | |
Squamous Ca ex PA | 1 | 3.5 | |
Adenocarcinoma NOS | 3 | 10 | |
Adenoid cystic carcinoma | 3 | 10 | |
Sebaceous carcinoma | 2 | 7 | |
Acinic cell carcinoma | 1 | <1 | |
Oncocytoma | 1 | <1 | |
Apocrine hidradenoma | 1 | <1 | |
Metastasis | 1 | – | 0.5 |
Melanoma skin | 1 | – |
Table 12.3
Histopathologic diagnoses of lacrimal sac lesions (N = 138)
Diagnosis (category) | N | Relative % | Absolute % |
|---|---|---|---|
Cysts | 1 | – | <1 |
Nasolacrimal duct | 1 | – | <1 |
Degenerations | 4 | 3 | |
Dacryolith | 2 | 1.5 | |
Fibrosis | 2 | 1.5 | |
Inflammation | 104 | 100 | 75 |
Dacryocystitis | 88 | 85 | 65 |
Infection | 3 | 3 | 2 |
Foreign bodygranuloma | 3 | 3 | 2 |
Lipogranuloma | 2 | 2 | 1.5 |
Granuloma | 2 | 2 | 1.5 |
Granulation tissue | 2 | 2 | 1.5 |
Lymphoid hyperplasia | 1 | 1 | <1 |
Sarcoidosis | 3 | 3 | 2 |
No diagnosis | 11 | 8 | |
Lymphoma | 2 | – | 1.5 |
DLBCL | 1 | – | |
B-CLL | 1 | – | |
Soft tissue | 6 | – | 4 |
Fibrous histiocytoma | 3 | – | 2 |
Hemangioma | 3 | – | 2 |
Epithelial | 10 | 100 | 7 |
Inverted papilloma | 2 | 20 | 1.5 |
Squamous cell carcinoma | 2 | 20 | 1.5 |
Transitional cell carcinoma | 2 | 20 | <1 |
Adenoid cystic carcinoma | 1 | 10 | <1 |
Adenocarcinoma NOS | 1 | 10 | <1 |
Adenocarcinomanasal sinus | 1 | 10 | <1 |
Melanoma (primary) | 1 | 10 | <1 |
12.2 Embryology, Anatomy, and Development
12.2.1 Embryology
A recent complete review of ocular and periocular embryogenesis has been provided in the second edition of the monograph by Barishak [1]. The organization of the orbit and its bony walls is governed by the optic cup and optic vesicle, which determine the morphogenesis of the orbital soft tissue contents and the bony orbital walls [2]. The embryonic and early fetal phase, during which one may speak of a “regio orbitalis,” is followed by a period of a primordial orbit which extends until after birth [3]. Unlike the trunk and extremities, the orbital bone and ocular connective tissue are derived from neural crest cells, not mesoderm. The connective tissue contributions of the neural crest are collectively referred to as mesectoderm. In the region of the brain rostral to the developing inner ear, the mesodermal segments are called somitomeres. The somitomeres give rise to the myoblasts of the extraocular muscles and vascular endothelium in and around the eye. It is important to point out that the mesenchyme is a broad term for any embryonic connective tissue, whereas mesoderm specifically relates to the middle embryonic layer. In the orbit, the mesenchyme consisting of fibrous and fibroadipose tissue, meninges of the optic nerve, sclera and episclera, vascular pericytes and striated extraocular muscle, satellite cells, peripheral nerve cellular elements, osteocytes, and cartilaginous elements are distinctive in being progeny of the mesectoderm [4, 5]. The migration of the neural crest cells proceeds over the face along two routes, which meet in the area of the orbit [6]. The maxillary wave of neural crest cells curves around the developing eye from below, while a frontonasal anlage migrates over the prosencephalon and the optic stalk from above [7]. Thus the floor and lateral wall of the orbit are contributed by the maxillary process, whereas the lacrimal and ethmoidal bones are contributed by the frontonasal process. Within the first 2 months of embryogenesis, the rudiments of the orbital bones are laid down. A unique mechanism of bone formation may be present in the maxillary and frontal bones of the orbit by way of ossification of the orbital muscle of Müller that forms the lateral and caudal walls of the embryonic orbit. In view of the developmental histology, the orbital muscle is a very unique smooth muscle mass in the human body: it connects between bones and is replaced by collagenous fibers even in fetuses. Moreover, the orbital muscle is likely to play a critical role as a septum to maintain a mechanical condition within the orbital space for the normal development of extraocular structures [8]. The lesser wing of the sphenoid bone is initially cartilaginous, but the greater wing and the rest of the orbital bones are membranous and ossify and fuse between the sixth and seventh months of gestation. The significance of this migratory pattern is important for understanding the location of congenital orbital, eyelid, and lacrimal anomalies. A failure of fusion between the neural crest waves results in clefting syndromes that involve the orbit. The typical location of the dermoid cysts at the frontozygomatic and frontomaxillary suture lines is the result of a sequestration of surface ectoderm in the areas of neural crest cell fusion. The superficial spread and deep invasion of basal cell carcinoma on the midface may be partially due to the location of the embryonic fusion planes. The orbital axes rotate from the early stages of optic cup development to adulthood. As the orbital bones develop, the eyes converge from an initial 180° position between the lateral orbital wall and the skull axis to 115° at birth, 68° in infancy, and their final position 45° in adulthood [9].
12.2.2 Anatomy and Histology
The orbits are pyramid-like or pear-shaped structures mainly limited by bony walls that protect and contain the eyeball and its adnexa. The stalks are the optic canals. The distance from the apex posteriorly to the orbital rim anteriorly varies from 40 to 50 mm, and the volume is 30 cm3. Seven bones comprise the orbital walls: frontal, greater and lesser wings of the sphenoid, ethmoid, palatine, maxilla, zygomatic, and lacrimal (Fig. 12.1). They separate the intraorbital tissues from the paranasal sinuses. Several apertures in the orbital bones allow the passage of blood vessels and nerves (Fig. 12.1). The orbital septum is a membranous sheet that acts as the anterior boundary of the orbit. It extends from the periosteum and periorbita at the orbital rim superiorly. It passes inferiorly into the eyelids to separate them from the orbit (preseptal and postseptal space) and fuses with the upper or lower eyelid retractors 2–4 mm from the tarsus (Fig. 12.2).
The contents of the orbit include the globe, optic nerve and meningeal sheaths, the extraocular striated muscles, levator palpebrae superior muscle and tendons, connective tissue fascia, and fat. Fibrocartilaginous tissue of the orbit is present in the trochlea of the tendon of the superior oblique muscle. Smooth muscle tissue is present in the remnants of the embryonal muscle of Müller. Connective tissue septa (the orbital reticulum) divide and connect orbital fat lobules and compartments [10–14]. A funnel-shaped muscle cone divides the orbit into an intraconal and extraconal space. The former is limited anteriorly by the posterior portion of the eye and includes the optic nerve, blood vessels, and nerves that supply the eye. A third “peripheral” or extraperiosteal space is defined as the potential space between the periosteum and the bony walls. Several central and peripheral nerves traverse the orbit. The ciliary ganglion, which transmits parasympathetic and sympathetic nerve fibers, is a 1.1-mm flat, oval structure that lies between the optic nerve and the lateral rectus muscle. The blood supply of the orbit mainly comes from the branches of the external and internal carotid arteries. The lateral orbit and forehead are supplied by the superficial temporal artery, a branch of the external carotid artery. The supraorbital and lacrimal arteries arise from the internal carotid arteries. The venous system of the orbit drains to the superior ophthalmic vein, which is joined by other veins coming from the eye and by the inferior ophthalmic vein at the orbital apex. The veins drain posteriorly into the cavernous sinus and inferiorly into the pterygoid plexus. There are no identifiable lymphatic vessels or lymph nodes in the bony orbit. The tissues from the most anterior part of the orbit drain into the lymphatics of the conjunctiva and eyelid.
12.3 Congenital Abnormalities
The spectrum of congenital abnormalities of the orbit includes choristomas, progonomas, hamartomas, and congenital malformations. Only choristomas and congenital malformations will be discussed in this section. For reasons of clarity, hamartomatous (most vascular) lesions will be discussed later in the section on vascular neoplasms. Progonoma is now considered a type of PNET and will be duly discussed in the neoplasm section.
12.3.1 Choristomas
12.3.1.1 Epithelial Cystic Choristoma
Definition
Dermoid, epidermoid, conjunctival, and respiratory cysts of the orbit are choristomas, i.e., cysts lined with mature epithelium that is not normally present in the orbit.
Epidemiology
Etiology
Epithelial cystic choristomas arise from a congenital rest of differentiated embryonic ectodermal tissue at the site of closure of a fetal cleft as described in the section on embryology.
Localization
Dermoid cysts are the most common cystic lesions and have a propensity for the superotemporal or supranasal anterior quadrants of the orbit.
Clinical Features
Typical is a superotemporal swelling of the orbit. CT scan demonstrates the bony relationship to the cystic lesion in 87 % of the cases, particularly at the frontal zygomatic suture. Rarely, these lesions may extend into the intracranial cavity through the bone. Deep-seated dermoids with bony involvement produce a characteristic punched-out appearance with hyperostotic margins, when the bone is viewed by x-ray and show a classical dumbbell sign on CT or MRI (Fig. 12.3).
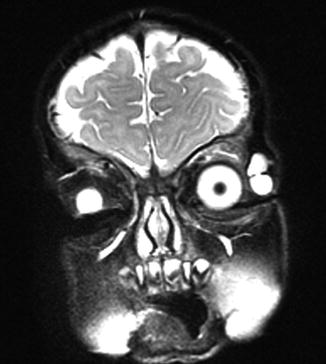
Fig. 12.3
MRI dermoid cyst: T2-weighted MR image showing a dumbbell-shaped lesion at the frontozygomatic fissure (Dr. Robert M. Verdijk)
Macroscopy
Dermoid cysts have a thin capsule and the lumen contains keratinous debris, hairs, and yellow lipid deposits. Dermoid cysts are usually spherical or oval in shape and range in size from 0.5 to 1.0 cm; an occasional lesion may reach 3 cm in size.
Histopathology
The lining of the dermoid cyst is composed of stratified squamous epithelium. Pilosebaceous units are observed in the cyst walls composed of dermal connective tissue (Fig. 12.4). When cysts rupture, the lining is partly replaced by a lipogranulomatous inflammatory response that extends into the adjacent orbital tissues. Rarely, the cyst wall does not contain adnexal structures (epidermoid cyst). Other rare cysts are lined by a double layer of cuboidal epithelium resembling conjunctiva (conjunctival cyst) or by ciliated cells (respiratory cyst). A combination of these is possible (Fig. 12.5).
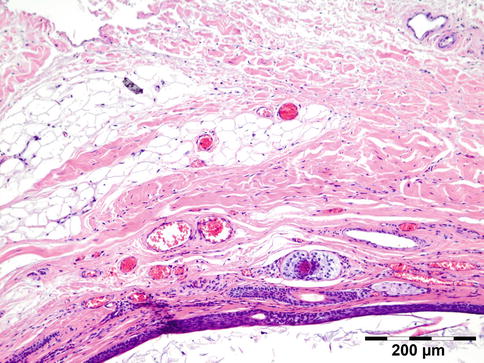
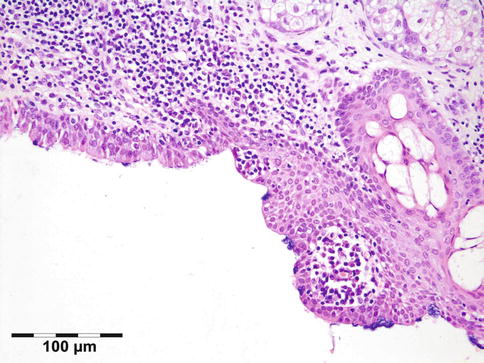
Fig. 12.4
Dermoid cyst: cyst wall lined by keratinizing epithelium with hair follicles and associated sebaceous glands in a dermis-like collagen and subcutaneous-like fat (HE 100×) (Dr. Robert M. Verdijk)
Fig. 12.5
Epithelial cystic choristoma: cyst wall partly lined by stratified squamous epithelium with goblet cells, partly lined by pseudostratified columnar epithelium with ciliated cells and goblet cells. Sebaceous glands indicating epidermal differentiation. Ciliated dermoid cyst (HE 200×) (Dr. Laura Bredow, Freiburg University; this case was presented at the 40th “Jahhrestagung der Deutschsprachigen Ophthalmopatologen,” Erlangen, 2012)
Differential Diagnosis
Traumatic conjunctival cysts may also show keratinization, but will not contain adnexal structures. Cholesterol granuloma does not show an epithelial lining. Mucocele of the paranasal sinus may also be lined with respiratory or squamous metaplastic epithelium. These lesions generally develop in older patients.
Histogenesis
The typical location of dermoid cysts at the frontozygomatic and frontomaxillary suture lines is the result of a sequestration of surface ectoderm in areas of neural crest cell fusion.
Genetics
Not applicable.
Prognosis and Predictive Factors
Some degree of rupture of the dermoid with granulomatous inflammation replacing the epithelium occurs in about 25 % of our cases of orbital dermoids. Rupture with accompanying inflammation and scarring may lead to larger, flat, or multilobulated lesions. The abrupt onset of manifestations may suggest the development of a malignant tumor. Complete resection of the intraosseous parts of the lesion is essential to prevent recurrence. Epithelial cystic choristomas have a very low capacity for malignant transformation to carcinoma.
12.3.1.2 Dermolipoma
These are not true orbital choristomas and will be described in the chapter on conjunctiva. Dermolipomas, however, frequently extend into the orbit and can be diagnosed as lipoma when prior presence of dermolipoma was not mentioned in the provided clinical information. We have diagnosed 4 cases in our series (<1 %).
12.3.1.3 Ectopic Lacrimal Gland (Cyst)
Definition
Lacrimal gland tissue in the deep orbital area outside and not in continuity with the lacrimal gland situated at the superotemporal quadrant.
Epidemiology
Rare.
Etiology
Sequestration of parts of the lacrimal anlagen.
Localization
Deep in the orbit or in the anterior quadrants other than the superior temporal quadrant.
Clinical Features
Although ectopic lacrimal glands of the orbit can be symptomatic at any age, they mostly present during the first three decades of life. Since the lesions are congenital, the reasons for their delayed manifestation are unclear [18].
Macroscopy
The specimen is composed of relatively normal-looking lacrimal gland tissue that may undergo cystic change to form a tense, thin-walled, clear fluid-filled cyst.
Histopathology
Microscopic examination shows normal-appearing lacrimal gland tissue, often with a moderate nonspecific inflammatory cell infiltration. Cystic cases show a cyst lined with cuboidal cells and occasional acini in the cyst wall (Fig. 12.6).
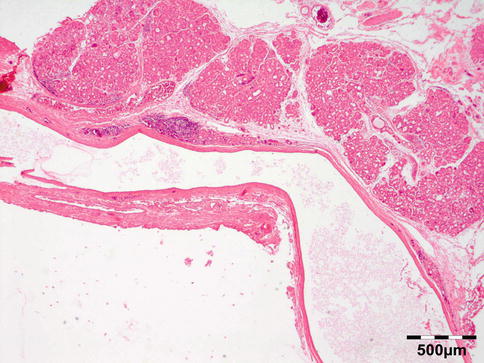
Fig. 12.6
Lacrimal gland cyst: cyst lined by cuboidal epithelium. Lacrimal gland tissue is present in the cyst wall (HE 25×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Clinical correlation is required to make this diagnosis since the normal lacrimal gland may extend as far as to the posterior part of the globe on the temporal side. The cuboidal lining ectopic lacrimal gland cyst may show resemblance to the lining of conjunctival cysts or dacryops.
Histogenesis
Not applicable.
Genetics
Not applicable.
Prognosis and Predictive Factors
Mostly the complications are inflammatory. Vascular lesions, pleomorphic adenoma, and very rare cases of carcinoma have developed in ectopic lacrimal gland tissue.
12.3.1.4 Osseous Choristoma
12.3.1.5 Ectopic Lymph Node
One case of an ectopic lymph node located in the lacrimal gland region has been reported [22].
12.3.1.6 Ectopic Pituitary Adenoma
One case of an ectopic pituitary adenoma in the orbit has been reported [23].
12.3.2 Teratoma
It has long been considered a choristoma but is now known to be a well-differentiated nonseminomatous germ cell tumor (NSGCT) and will be discussed in the section on neoplasms.
12.3.3 Hamartoma
Hamartomas are proliferations of disorganized tissues that are encountered in their normal location. Most vascular lesions will be discussed in the section on vascular neoplasms
12.3.3.1 Varix
Definition
A type of vascular aneurysm in the orbit. Orbital varices are a vascular hamartoma typified by a plexus of low-pressure, low-flow, thin-walled, and distensible vessels that intermingle with the normal orbital vessels [24].
Epidemiology
Orbital varix is a rare entity, accounting for less than 1 % of all orbital lesions in our series. Orbital varix represented 10 % of all vascular orbital lesions. Although it is believed to be congenital, and thus present at birth, patients typically do not become symptomatic until later childhood or early adulthood (10–30 years of age). Cases have however been reported at essentially any age [25].
Etiology
Orbital venous varices are divided into primary and secondary. Primary orbital varices are idiopathic and most likely congenital. They are confined to the orbit. Secondary orbital venous varices are those that are acquired due to increased blood flow as a result of intracranial arteriovenous malformations, caroticocavernous fistula, and dural arteriovenous fistula, which drain via the orbit [25, 26].
Localization
Mostly a massively enlarged ophthalmic vein is located in the superior orbit. Most of the lesions are unilateral, but rarely may they be bilateral.
Clinical Features
Evanescent unilateral proptosis is the principal sign of orbital varix. In 20 % of the cases, Phleboliths may form in association with varix and may aid in radiological diagnosis [24].
Macroscopy
The varix has features of a thin-walled vessel with a large lumen that may show phleboliths.
Histopathology
Smooth muscle cells of the media are often decreased in numbers. In the lumen a phlebolith may be present.
Differential Diagnosis
Hemangioma, lymphangioma, idiopathic orbital inflammation (IOI), carotid cavernous fistula, and dural shunt.
Histogenesis
The ophthalmic vein is of mesodermal origin.
Genetics
No genetic abnormalities have been reported.
Prognosis and Predictive Factors
Most orbital varices may be managed conservatively and only warrant surgery in the presence of recurrent thrombosis, disfiguring proptosis, or acute visual loss. Alternatively endovascular coiling has been reported in selected cases.
12.3.4 Congenital Malformations
12.3.4.1 Microphthalmos with Cyst
Isolated microphthalmia associated with colobomatous cyst results from a defect in the closure of the embryonic fissure at the 7- to 20-mm stage of development. Microphthalmia can be associated with either a small, clinically undetectable cyst or a large, typically inferior cyst that deforms the eye and its surroundings. It is usually unilateral, although bilateral cases have been described [27] (see also page 20).
12.3.4.2 Meningoencephalocele and Ectopic Brain Tissue
Definition
Saclike herniations of the brain that protrude into the orbit.
Synonyms
Not true synonyms, but depending on contents, these lesions are designated meningocele, encephalocele, or meningoencephalocele (see below).
Epidemiology
Rare.
Etiology
The cephalocele probably is a cleavage disorder where some portion is separated from the neurectoderm by subsequent bone development. A true cephalocele should have some communication with the brain cavity; otherwise, one should classify the lesion as ectopic brain tissue. Only very rare cases of ectopic brain tissue without connection to the brain have been reported. These probably represent a completely sequestrated herniation [28]. Other theories on ectopic brain tissue include neuroectodermal rests with a potential for differentiation, preferential development of one germ layer of a teratoma into the neural cell lines, and a true astrocytoma.
Localization
These herniations may be classified according to position or contents. The latter classification can only be determined by histologic examination as meningocele, encephalocele, or meningoencephalocele [15]. The former classification distinguishes anterior (frontoethmoidal) or posterior encephaloceles.
Clinical Features
Broad nasal bridge. Orbital swelling. Associations are with microphthalmia, orbital varices, colobomas, and morning glory syndrome [29].
Macroscopy
Meningoencephaloceles consist of jumbled often highly vascularized fragments of glossy white brain tissue and arachnoid.
Histopathology
Microscopic examination may show a dense fibrous wall with neural- and arachnoid-like tissue (Fig. 12.7).
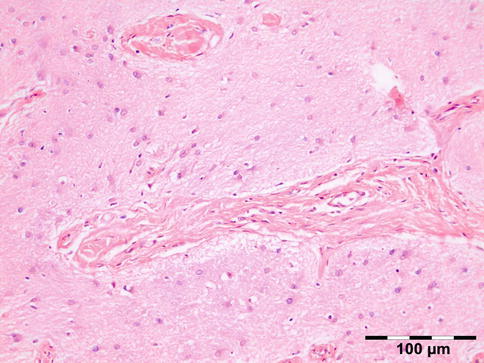
Fig. 12.7
Encephalocele: glial tissue with astrocytes, oligodendroglial cells, and microglia with interspersed fibrous collagen-rich strands (HE 200×) (Dr. Jose G. Cunha Vaz; this case was presented at the EOPS meeting, Munich, 1977, case 77–41)
Differential Diagnosis
Microphthalmos with cyst.
Histogenesis
Not applicable.
Genetics
Neurofibromatosis is often associated with posterior encephaloceles.
Prognosis and Predictive Factors
Pre- or postoperative liquor leakage may occur and should be addressed since this may offer a potential route for infectious agents into the brain cavity.
12.4 Inflammatory Diseases
Orbital inflammatory diseases are mass-forming lesions occurring in patients of various ages. The spectrum ranges from disease caused by infection, surgical procedures, trauma, and foreign bodies to nonspecific inflammation caused by immune reactions and local disease or secondary to an underlying systemic inflammatory disease and myofibroblastic neoplasms [30].
12.4.1 Infectious
Microorganisms: Acute/chronic/granulomatous inflammation may be caused by a host of microorganisms such as bacteria (e.g., S. pneumoniae, S. aureus, S. pyogenes, H. influenzae, Actinomyces, Mycobacterium tuberculosis (Fig. 12.8)), fungi (Aspergillus fumigatus (Fig. 12.9), mucormycosis (Fig. 12.10)), maggots (myiasis), and parasites (helminths; see below). In many cases infectious disease is related to extension of infection from adjacent areas such as preseptal fat, nasal cavities, or the cranial cavity. Orbital cellulitis may develop after a penetrating trauma. Osteomyelitis may present as an orbital infection (Fig. 12.11). Infections represented 10 % of all inflammatory lesions in our series. Some cases may be related to immune-compromised state due to either diabetes mellitus, HIV/AIDS, or immunosuppressive therapies for autoimmune disease, chemotherapy for malignancy, stem cell transplantation, or solid organ transplantation. When infectious disease is suspected, a combination of histochemical stains like Gram, Giemsa, Grocott, PAS, alcian blue, and Ziehl–Neelsen should be performed in order to increase the chances of identification of the microorganisms. Immunohistochemistry may aid the identification of Toxoplasma, Treponema pallidum, HSV, VZV, or EBV when indicated.
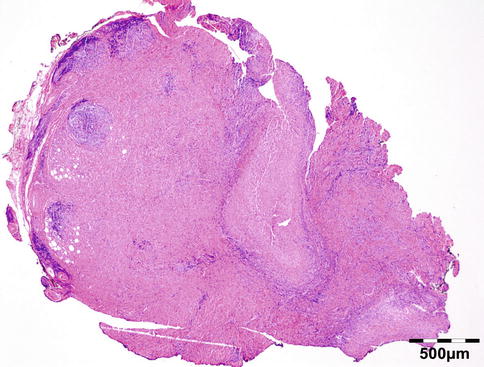
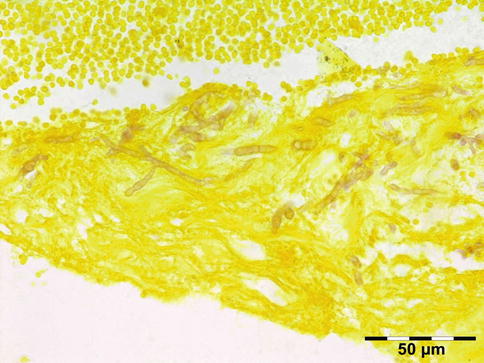
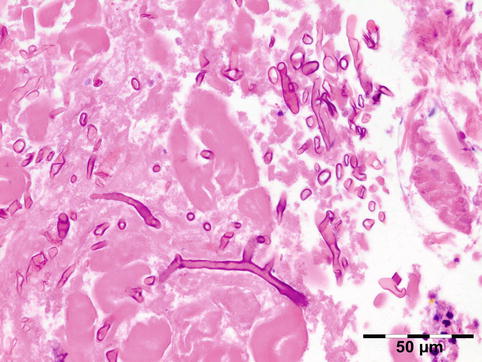
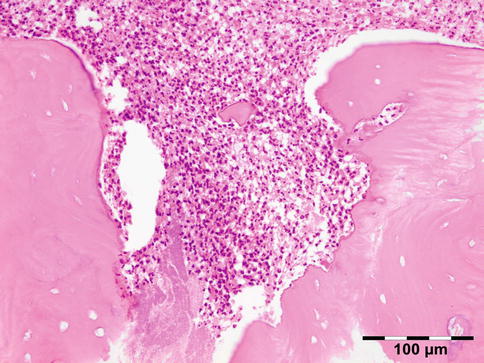
Fig. 12.8
Tuberculosis: necrotizing granuloma. Mycobacterium tuberculosis could only be demonstrated by PCR. The patient showed a good response to tuberculostatic therapy (HE 25×) (Dr. Robert M. Verdijk)
Fig. 12.9
Aspergillus: septated hyphae showing branching at acute angles, often 45° fit the diagnosis of Aspergillus (Grocott 400×) (Dr. Pierre Danis; this case was presented at the EOPS meeting, Toulouse, 1974, case 74–31)
Fig. 12.10
Mucormycosis: broad nonseptated hyphae showing branching at 90° angle fit the diagnosis of Mucormycosis. The cell walls are typically well delineated in HE sections (HE 400×) (Dr. Robert M. Verdijk)
Fig. 12.11
Osteomyelitis: nonvital bony sequesters are seen in an acute inflammatory background with neutrophils. Note the loss of chromatin staining in the osteocytes of the lamellar bone (HE 200×) (Dr. Robert M. Verdijk)
Helminths: Man may become infected by three types of larval cysts of tapeworms: (1) the hydatid of Echinococcus granulosus, a larval cyst of variable size in which daughter cysts develop; (2) coenurus, a larger single bladder worm of Taenia multiceps, 5 cm or more in diameter, containing several hundreds of scoleces; and (3) Cysticercus cellulosae, a small bladder worm of Taenia solium, containing one single scolex [31]. The adult worms of Onchocerca on the other hand may cause a tumor known as onchocercoma.
12.4.1.1 Hydatid Cyst
Definition
Infestation with the larval cyst of Echinococcus granulosus.
Epidemiology
Echinococcus granulosus has a worldwide distribution, mainly in sheep- and cattle-raising countries. The definitive hosts are dogs or other canids.
Etiology
Humans become infected by ingesting eggs on fomites or in food and water contaminated with dog feces.
Localization
Clinical Features
The patient may present with a variety of symptoms and signs, including painless proptosis, oculomotor palsies, and eyelid edema. On MRI orbital hydatid cysts usually appear as a well-defined, thin-walled, oval-shaped lesions with fine peripheral rim enhancement of their fibrous capsule after contrast medium administration.
Macroscopy
The cysts are typically white, subspherical in shape, and fluid filled.
Histopathology
The cyst wall consists of an inner nucleated germinal layer of epithelium and an opaque, elastic acellular laminated layer of varying thickness that is surrounded externally by a host-derived fibrous layer of tissue (Fig. 12.12). Brood capsules develop from the germinal layer and mature to form protoscoleces (Fig. 12.13). Each scolex is about 100 μm across and contains suckers, a double crown of hooklets (Fig. 12.14), and calcareous bodies. Daughter cysts are usually produced within the mother cavity. Death and degeneration of free brood capsules results in protoscoleces in the fluid contents, referred to as hydatid sand.
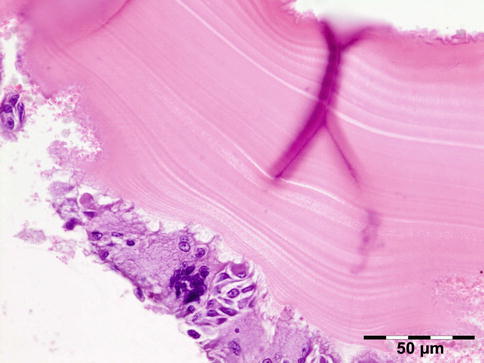
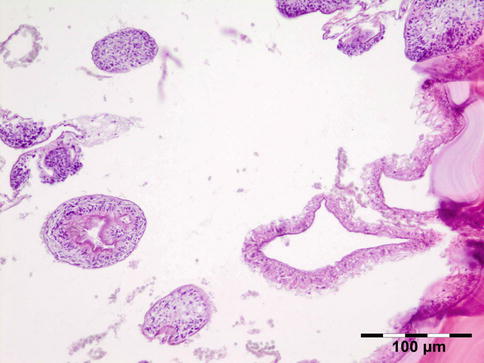
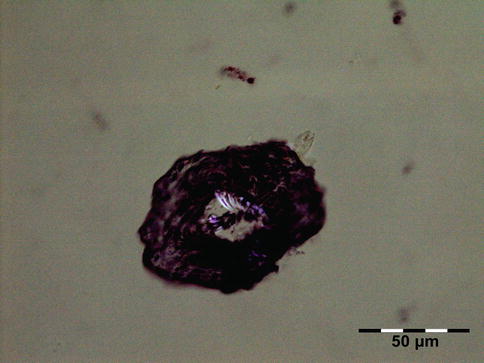
Fig. 12.12
Hydatid cyst: typical lamellated outer cyst wall surrounded by inflammatory cells including multinucleated giant cells (PAS 400×) (Dr. Martine Brihaye-van Geertruyden; this case was presented at the EOPS meeting, Rotterdam, 1967, case 67–25)
Fig. 12.13
Hydatid cyst: inner cyst wall with a nucleated germinal layer of epithelium. Brood capsules attached to the germinal layer and free floating daughter cysts can be seen (PAS 200×) (Dr. Martine Brihaye-van Geertruyden)
Fig. 12.14
Hydatid cyst: birefringence easily identifies protoscoleces (polarized light 200×) (Dr. Martine Brihaye-van Geertruyden)
Differential Diagnosis
Coenurus and cysticercosis.
Histogenesis
Not applicable.
Genetics
Not applicable.
Prognosis and Predictive Factors
Most patients with orbital parasitic cysts have recovered reasonably good vision following surgical extirpation of the mass or antihelminthic medication.
12.4.1.2 Coenurus
Definition
Infestation with coenuri of Taenia multiceps.
Epidemiology
Taenia multiceps distribution is worldwide.
Etiology
Its most common definite host is the domestic dog, or other canids. Humans become infected in the same way as described for Echinococcus.
Localization
After about 3 months, coenuri of T. multiceps develop in the central nervous system preferably, especially in the cerebrospinal fluid pathways, and in the eye and orbit [31].
Clinical Features
Redness of the eye and cystic tumor.
Macroscopy
A coenurus is a white or gray spherical or ovoid unilocular cyst which varies from a few millimeters to 2 cm in diameter.
Histopathology
The cyst wall is surrounded by a thin layer of host-derived fibrous tissue that often shows minimal inflammatory reaction. However, the degenerating coenurus may be heavily infiltrated by mononuclear and polymorphonuclear inflammatory cells with a great number of eosinophils (Fig. 12.15). Inside the fibrous wall, the wall of a cystic larva consists of a thin outer cuticular layer (the tegument) 5 μm thick, covered with microvilli. The inner cellular layer contains smooth muscle fibers lined by a row of tegumental cells and parenchyma (Fig. 12.16). The circular cavity is filled with fluid. The inner wall has a linearly arranged large number of inverted scoleces. Each scolex contains a rostellum with a double row of small hooks of two sizes. Everted scoleces may be present.
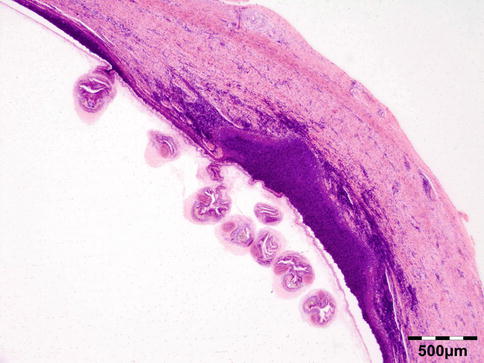
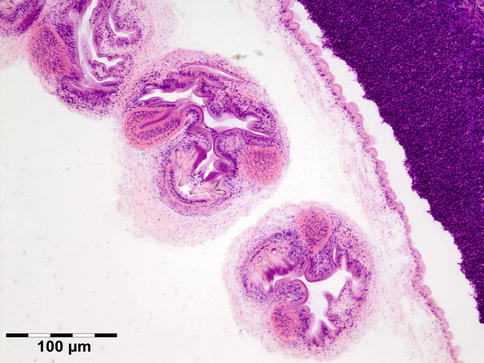
Fig. 12.15
Coenurus: the cyst wall is surrounded by a heavy mononuclear and polymorphonuclear infiltrate. The circular cavity is filled by fluid. The inner wall has a linearly arranged large number of inverted and everted scoleces (HE 25×) (Dr. Robert M. Verdijk)
Fig. 12.16
Coenurus: the inner cellular layer with smooth muscle fibers is lined by a row of tegumental cells and parenchyma. Each scolex contains a rostellum with a double row of small hooks of two sizes (HE 200×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Hydatid cyst and cysticercosis.
Histogenesis
Not applicable.
Genetics
Not applicable.
Prognosis and Predictive Factors
Most patients with orbital parasitic cysts have recovered reasonably good vision following surgical extirpation of the mass or antihelminthic medication.
12.4.1.3 Cysticercosis
Definition
Infestation with the cystic larval form of the swine tapeworm Taenia solium.
Epidemiology
T. solium is cosmopolitan in spread. Orbital and adnexal cysticercosis is now considered a common disease in endemic regions such as Mexico, Africa, India, China, Indonesia, Eastern Europe, and South and Central America [33].
Etiology
Cysticercosis is caused by hematogenous spread and encystment of the larval form of the swine tapeworm Taenia solium. The worm passes its life cycle in two hosts. Humans are the definitive hosts and the adult parasites live in the small intestine for several years. The pig is the intermediate host and the main host of the larva. Man acquires infection through the ingestion of infective cysts present in contaminated food such as undercooked pork.
Localization
The parasites reach the bloodstream via the intestine and then undergo passive hematogenous spread to other tissues and organs, tending to embed themselves in areas with a high metabolic turnover and good glycogen supply such as the central nervous system, eye, and striated muscles [34]. As a definitive host, man harbors the adult tapeworm, and as an intermediate host, man harbors the bladder worm or cysticercus.
Clinical Features
The various clinical presentations of cysticercosis are redness of the eye, proptosis, restriction of ocular motility, and cystic tumor.
Macroscopy
Cysticerci are spherical to oval milky white cysts about 1 cm in diameter when fully developed.
Histology
The cyst contains fluid and a single invaginated scolex (Fig. 12.17). The scolex has four large suckers and a rostellum with a double row of large and small hooklets. The cyst wall is 100–200 μm thick and often is raised into projections that are 10–25 μm in diameter. The tegument is usually less than 5 μm and its outer surface is covered with microvilli. Numerous smooth muscle fibers lie immediately beneath the tegument and extend into the parenchyma. A row of tegumental cell lies beneath the muscle fibers. The cysticercus is surrounded by fibrous tissue and often causes virtually no inflammatory response. However, when cysticerci degenerate, there is an infiltration of neutrophils, histiocytes, and eosinophils. Eventually a granulomatous reaction may occur characterized by histiocytes, epithelioid cells, and foreign body giant cells, leading to fibrosis and calcification.
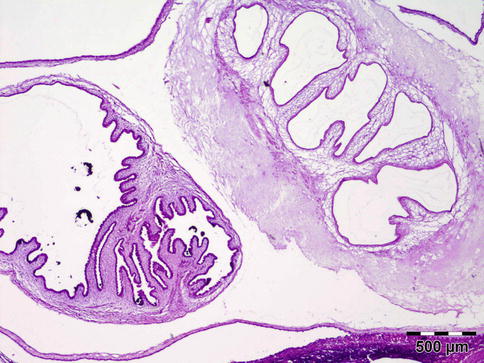
Fig. 12.17
Cysticercoma: a cyst containing fluid. The single invaginated scolex often cannot be found in histology sections. The cyst wall is raised into projections. The cysticercus is surrounded by fibrous tissue and caused virtually no inflammatory response (HE 25×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Cysticercosis can masquerade as any of the subacute, acute, or chronic inflammatory or cystic lesions of the orbit. Hydatid cyst, coenurus.
Histogenesis
Not applicable.
Genetics
Not applicable.
Prognosis and Predictive Factors
Surgical removal of extraocular muscle cysts is usually contraindicated. Treatment with antihelminthic drugs under steroid cover has a high success rate in cyst elimination.
12.4.1.4 Onchocercoma
Definition
Infestation with the adult worm of Onchocerca volvulus.
Epidemiology
Onchocerca volvulus is commonly found in West, Central, and East Africa, in South America, and less commonly in the Middle East.
Etiology
Humans are the only definitive host for O. volvulus. The intermediate host or vector is the black fly (Simulium). The parasite is acquired through the bite of an infected fly.
Localization
The adult worm of Onchocerca is most often seen in the subcutaneous tissues and conjunctiva but may extend into the anterior orbit.
Clinical Features
It is the cause of river blindness in man; the orbit may be affected by onchocercomas which are palpable nodi that may occur adherent to the periosteum of the orbital bones [35].
Macroscopy
Nodular fibrous tissue, sometimes the outline of the worms can be observed on cut surface.
Histopathology
Histologically the nodule consists of three zones (Fig. 12.18): an external poorly vascularized, fibrous capsule, an intermediate fibrocellular layer consisting mainly of granulation tissue, and a central zone which, depending on the age of the nodule, may contain adult worms (Fig. 12.19) and microfilariae, or the caseous remnants of dead worms.
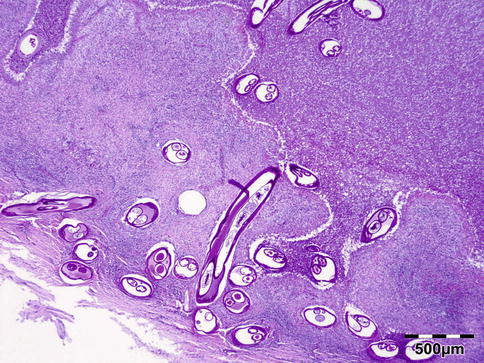
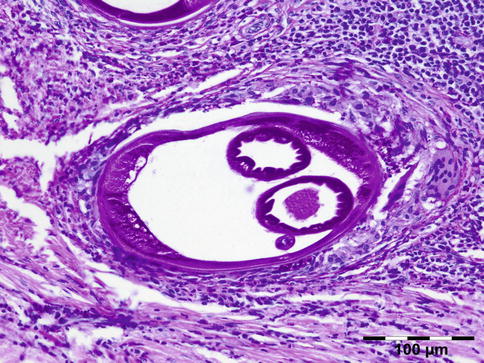
Fig. 12.18
Onchocercoma: a fibrous capsule surrounds granulation tissue with granuloma formation and adult worms (PAS 25×) (Dr. Robert M. Verdijk)
Fig. 12.19
Onchocercoma: adult female worm surrounded by a dense inflammatory infiltrate containing multinucleated giant cells. The cuticle shows ridges. Inside the worm muscle cells, gut, and uterus can be seen (PAS 200×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Other infectious diseases of the orbit.
Histogenesis
Not applicable.
Genetics
Not applicable.
Prognosis and Predictive Factors
Antihelminthic drugs as ivermectin are effective in eradicating the worms.
12.4.2 Reactive Inflammation
12.4.2.1 Cholesterol Granuloma
Definition
Foreign body reaction against cholesterol crystals.
Synonyms
The term “cholesteatoma” has incorrectly been used for any lesion containing cholesterol crystals and should be avoided.
Epidemiology
A rare entity.
Etiology
Cholesterol granuloma of the orbit has been associated with a previous trauma. They are relatively rare and represented only 1 % of all orbital lesions in our series. Approximately 50 % of the cases of orbital cholesterol granulomas, however, are not associated with previous trauma. Thus, pathogenesis of cholesterol granuloma still remains unclear. It has been suggested that cholesterol is produced as a result of the breakdown of blood components, and this is followed by foreign body granuloma formation. The adjacent bony resorption is believed to be initiated by the prostaglandins produced by the platelets within the hematoma.
Localization
CT usually shows an isodense to hypodense osteolytic lesion in the superolateral bony orbit with an extraconal soft tissue mass, leading to inferior and anterior displacement of the globe.
Clinical Features
Typically, orbital cholesterol granulomas have a male predilection and occur between the ages of 30 and 60 years [36]. Orbital cholesterol granulomas have characteristic radiologic imaging findings.
Macroscopy
Reddish-brown granular material may be accompanied by thick, reddish-brown fluid.
Histopathology
Shows numerous slit-like spaces that represent cholesterol crystals surrounded by multinucleated foreign body-type giant cells, lipid-laden histiocytes, and clusters of hemosiderin deposits or hematoidin crystals (Fig. 12.20). Focal areas of calcified bone trabeculae may be noted. Scarce fibrous, amorphous, and necrotic areas may also be present.
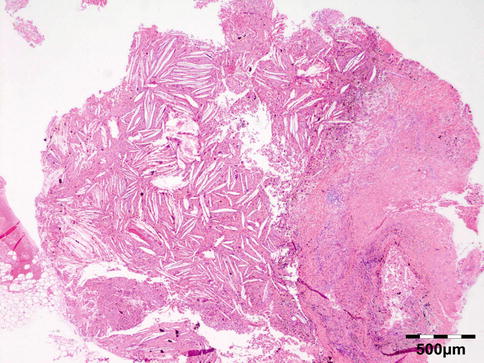
Fig. 12.20
Cholesterol granuloma: slit-like spaces that represent cholesterol crystals are surrounded by multinucleated foreign body-type giant cells, lipid-laden histiocytes, and clusters of hemosiderin deposits or hematoidin crystals. Focal areas of dystrophic calcification are present within amorphous and necrotic areas (HE 25×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Although the histopathological features of epidermoid cysts (cholesteatoma) and cholesterol granuloma can be very similar, the differentiating histopathological feature is the presence of epithelial elements in epidermoid or cholesteatoma and their absence in cholesterol granulomas [37]. Giant cell reparative granuloma of bone.
Histogenesis
Not applicable.
Genetics
Not applicable.
Prognosis and Predictive Factors
May recur after surgery.
12.4.2.2 Paraffinoma/Sclerosing Lipogranuloma
Definition
Lipogranulomatous inflammation reactive to exogenous paraffin (Fig. 12.21), silicone oil (used in retinal detachment) [38], Vaseline ointment containing antibiotics/ointment plug (after endonasal surgery) [39], or hydrogel (Fig. 12.22).
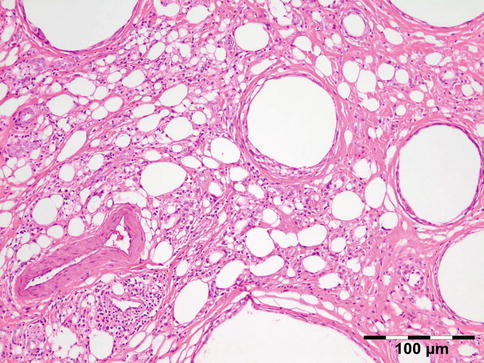
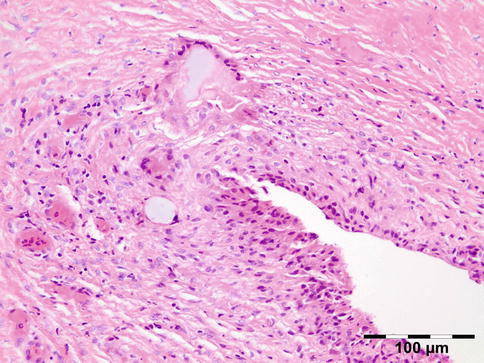
Fig. 12.21
Paraffinoma: a xanthogranulomatous inflammation with interstitial vacuolar clear rounded spaces with varying diameter surrounded by foamy macrophages, a “Swiss cheese pattern” (HE 200×) (Dr. Robert M. Verdijk)
Fig. 12.22
Hydrogel granuloma: granulomatous inflammation with slit-like spaces and multinucleated macrophages filled with basophilic amorphous material (HE 200×) (Dr. Robert M. Verdijk)
Epidemiology
Rare; paraffinoma represented only 1 % of all orbital lesions in our series and only 2.5 % of all inflammatory lesions.
Etiology
May be the result of surgical procedures or medical treatment but may also be observed as self-inflicted lesions such as unprofessional corrective introduction of exogenous materials into the tissues.
Localization
Extraconal.
Clinical Features
The symptoms are of a space-occupying inflammatory lesion.
Macroscopy
Normal pink to yellowish tissue may be observed.
Histopathology
The xanthogranulomatous inflammation exists as interstitial vacuolar clear rounded spaces with varying diameter surrounded by foamy macrophages giving the appearance of a “Swiss cheese pattern.” In the case of ointment plugs, pigment of iodine may be observed. In older lesions the single granulomas consisting of tightly packed epithelioid cells may be separated by small strands of collagenous tissue containing vessels and lymph follicles, sometimes with germinal centers. The longer the granulomas exist, the more the vacuoles decrease in number while strands of dense connective tissue predominate, surrounding small islands of epithelioid histiocytes. Larger vacuoles are replaced by concentric layers of hyaline tissue.
Differential Diagnosis
Xanthoma, xanthogranuloma.
Histogenesis
Not applicable.
Genetics
Not applicable.
Prognosis and Predictive Factors
Though not a common event, postoperative paraffin granuloma is a severe complication leading to chronic and recurrent disease with considerable functional impairment. The use of paraffin-containing ointment in sinus surgery should be abandoned, especially if peri- or postoperative bleeding occurs.
12.4.3 Immune Reactive Diseases
12.4.3.1 Thyroid Eye Disease
Definition
Graves’ disease is an autoimmune inflammatory disorder, caused by thyroid autoantibodies resulting in hyperthyroidism.
Epidemiology
About 30–50 % of patients with thyroid dysfunction will also suffer from Graves’ orbitopathy. Thyroid eye disease usually presents during midlife but may occur in children, adolescents, and the elderly. These samples are rarely submitted for histopathological examination and represented only 1.5 % of all inflammatory lesions in our series.
Etiology
Inflammatory cells in the orbit produce factors activating orbital fibroblasts that probably play a key role in the pathogenesis of thyroid eye disease [40].
Localization
Involves the interstitial tissues of the extraocular muscles, most commonly the inferior rectus and medial rectus muscles. Also the orbital and preseptal fat may be involved.
Clinical Features
Swelling of the eye muscles, resulting in upper eyelid retraction, redness, conjunctivitis, and proptosis [41].
Macroscopy
Firm white edematous tissue fragments.
Histopathology
Histologic evaluation shows an increase of glycosaminoglycans (largely hyaluronic acid) that stain positive for alcian blue and collagen staining positive for Sirius red. A cellular infiltration may show admixed lymphocytes, plasma cells, macrophages, and mast cells (Fig. 12.23).
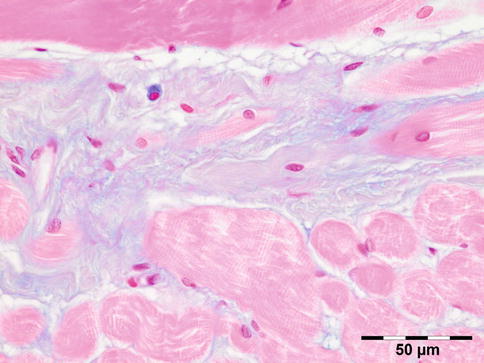
Fig. 12.23
Graves’ orbitopathy: striated muscle fibers are separated by a widened interstitium with increased glycosaminoglycans that stain positive for alcian blue. An occasional mucin-stained macrophage is present (alcian blue 400×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Infection, IOI.
Histogenesis
Fibroblasts and inflammatory cells are mesodermal-derived tissue. Tentatively, it might be that because the extraocular muscles are of mesectodermal rather than of mesodermal origin, they may be immunologically distinct from skeletal muscles.
Genetics
No genetic abnormalities have been reported.
Prognosis and Predictive Factors
Patients who fail to improve on medical or radiotherapeutic treatment may improve on decompressive surgery.
12.4.3.2 Inflammatory Pseudotumors
Idiopathic Orbital Inflammatory (IOI) Disease
Definition
A space-occupying inflammatory disorder that simulates a neoplasm but has no recognizable cause.
Synonyms
Idiopathic inflammatory pseudotumor (IIP), orbital pseudotumor. When restricted to the extraocular muscles, this condition may be called orbital myositis; when restricted to the lacrimal gland, it is frequently called (sclerosing) dacryoadenitis.
Epidemiology
IOI is one of the most common acute, painful orbital masses in the adult population. IOI is the second most common cause of exophthalmos following Grave’s orbitopathy [42] and the third most common orbital disorder following thyroid orbitopathy and lymphoproliferative disease, accounting for 5–17.6 % of orbital disorders [43, 44]. IOI represented 8.5 % of all orbital lesions in our series and 29.5 % of all inflammatory lesions. There is no age, sex, or race predilection, but it is most frequently seen in middle-aged individuals. Pediatric cases account for 17 % of all cases of IOI [45].
Etiology
The exact etiology of IOI is unknown, but infectious and immune-mediated mechanisms have been postulated. IOI has also been observed in association with Crohn’s disease, systemic lupus erythematosus, rheumatoid arthritis, diabetes mellitus, myasthenia gravis, and ankylosing spondylitis, all of which strengthen the basis of IOI being an immune-mediated disease. Response to corticosteroid treatment and immunosuppressive agents also supports this idea [43].
Localization
IOI can range from a diffuse inflammatory process to a more localized inflammation of the extraocular muscles (myositis), the adjacent adipose tissue, or the lacrimal gland (sclerosing dacryoadenitis).
Clinical Features
The symptoms are those of a space-occupying mass, associated with proptosis, and cranial nerve palsy (Tolosa–Hunt syndrome). The diagnosis is most commonly made on a clinical basis; however, in atypical cases orbital biopsy is required to exclude other orbitopathies [46]. IOI is a diagnosis of exclusion, both clinically and histopathologically.
Macroscopy
Gray-white solid mass with a variable aspect of fibrosis.
Histopathology
IOI is a nongranulomatous inflammation of lymphocytes, plasma cells, eosinophilic granulocytes, and often lymphoid follicles. The stroma may vary from edematous to fibrotic/sclerotic. It has been shown that the sclerosing subtype has a poorer response to steroid treatment (Fig. 12.24) [47].
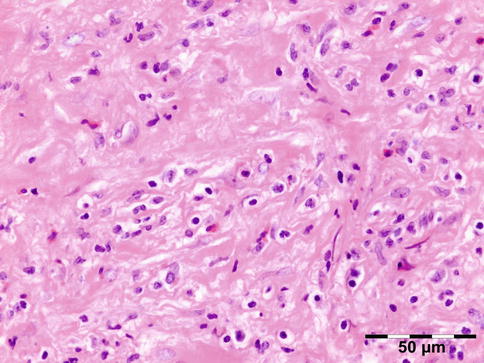
Fig. 12.24
Idiopathic orbital inflammation (IOI): inflammation of lymphocytes, plasma cells, eosinophilic granulocytes, and histiocytes. The stroma is severely fibrotic/sclerotic with an increase in collagen fibers (HE 400×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Specific inflammatory reaction patterns such as multinucleated giant cells, granuloma, xanthogranuloma, vasculitis, necrosis, or necrobiosis should not be present.
Genetics
Oligoclonal or pseudomonoclonal B- or T-cell populations may be demonstrable, which however by itself does not provide proof of a lymphoma.
Prognosis and Predictive Factors
Response to corticosteroid treatment supports the postulate of immune-mediated disease [43]. Therapeutic modalities included observation alone, antibiotics, oral corticosteroids, intravenous corticosteroids, adjunctive radiation therapy, and systemic immunosuppressive drugs.
IG4-Related Orbital Disease (IGRD)
Definition
IGRD is a recently defined inflammatory process characterized by tissue infiltration by IgG4-bearing plasma cells.
Epidemiology
IGRD affects males and females equally which is in contrast to IGRD pancreatitis [48]; the mean age of onset is 55 years.
Etiology
The pathogenesis of IgG4-related disease (IGRD) is poorly understood; findings consistent with both an autoimmune disorder and an allergic disorder are present [49].
Localization
Classical sites of involvement include the pancreas, hepatobiliary tract, salivary gland, lymph nodes, and retroperitoneum. IgG4-associated disease is now recognized as a systemic process. IGRD may also affect the lacrimal glands and periocular tissues [50]. IGRD also appears to account for 25–50 % of orbital pseudotumors and is additionally recognized now as a cause of orbital myositis (IgG4-related orbital myositis) [51]. The eyelid, lacrimal gland, extraocular muscle, orbital soft tissue, and nasolacrimal duct may all be affected. The conjunctiva has not been reported; the lacrimal gland is most common [49, 52–54]. Simultaneous involvement of two pairs of lacrimal glands, submandibular glands, or parotid glands is known as Mikulicz’s disease.
Clinical Features
Macroscopy
Gray-white solid mass with a variable aspect of fibrosis.
Histopathology
IGRD is characterized by varying degrees of focal or diffuse fibrosis and IgG4-positive-rich lymphoplasmacytic infiltration, with subsets of pseudolymphomatous, mixed, or xanthogranulomatous pattern (Figs. 12.25 and 12.26) [56, 57]. For the histologic diagnosis of IGRD, the areas of maximum IgG4-positive staining should be selected [48]. The number of IgG4-positive plasma cells per high-power field (HPF) regarded as sufficient varies somewhat from tissue to tissue. Generally, the minimum for making the diagnosis for most tissues is from 30 to 50 IgG4-positive cells/HPF. The ratio of IgG4+/IgG+ should excess 0.4 (Figs. 12.27 and 12.28) [48]. Obliterative phlebitis is not a common feature in orbital IGRD [49].
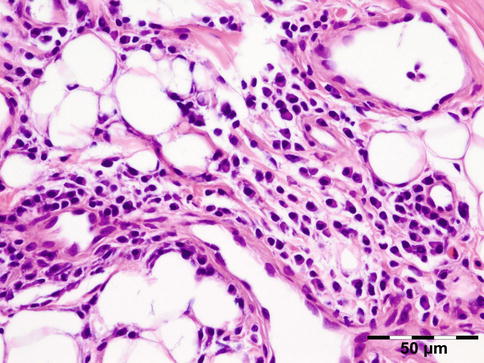
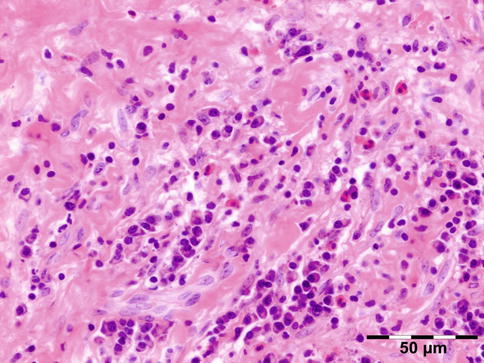
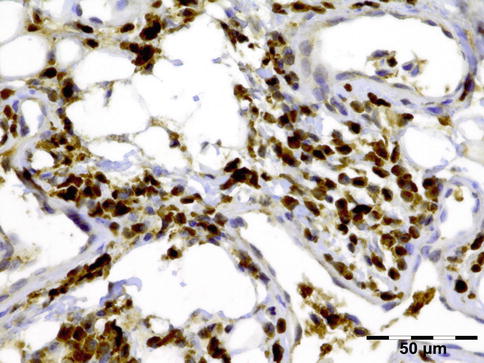
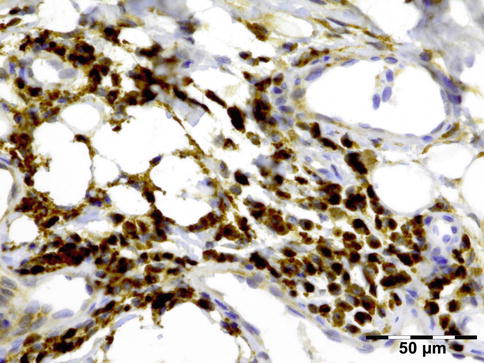
Fig. 12.25
IgG4-related orbital disease: the early phase of the disease shows a rich lymphoplasmacytic infiltration with perivascular involvement and a slight increase in collagen fibers (HE 400×) (Dr. Robert M. Verdijk)
Fig. 12.26
IgG4-related orbital disease: the biopsy of a late recurrence (same patient as Fig. 12.25) after 20 years shows a severely sclerotic lesion with a rich lymphoplasmacytic infiltration with admixed eosinophils. Phlebitis or vasculitis is not always present in orbital disease (HE 400×) (Dr. Robert M. Verdijk)
Fig. 12.27
IgG4-related orbital disease: immunohistochemical staining for IgG shows an increased amount of IgG-positive plasma cells of over 50/HPF (IgG IH 400×) (Dr. Robert M. Verdijk)
Fig. 12.28
IgG4-related orbital disease: immunohistochemical staining for IgG4 shows a clearly increased amount of IgG-positive plasma cells of over 50/HPF in a high ratio when compared to IgG (IgG4 IH 400×) (Dr. Robert M. Verdijk)
Differential Diagnosis
IOI, Wegener’s granulomatosis.
Genetics
No genetic abnormalities have been reported.
Prognosis and Predictive Factors
A good initial therapeutic response to glucocorticoids is characteristic. Patients may improve spontaneously without treatment, but many relapse in time. Sustained benefit may be observed in treated patients, but relapses are common after discontinuation of therapy. Additional organs and tissues may become involved over time, sometimes despite apparently effective treatment. Extranodal marginal zone B-cell lymphoma of the mucosa-associated lymphoid tissue may be associated with IGRD [58].
Xanthogranulomatous Inflammatory Diseases
Definition
A disease characterized by histiocytes, Touton giant cells, and lymphocytic inflammation.
Orbital xanthogranulomatous disease constitutes a group of entities with varying manifestations that are poorly understood. The clinical presentation in adults consists of uni- or bilateral orbital mass lesions or periocular yellow-brown infiltrates that may mimic xanthelasmata. These abnormalities tend to progress, causing eyelid disfigurement, eyeball displacement, eyeball motility disturbances, and – rarely – optic neuropathy. Orbital xanthogranulomatous disease is now considered to constitute a spectrum of four entities: adult-onset xanthogranuloma (AOX) (solitary lesion without systemic findings), adult-onset asthma and periocular xanthogranuloma (AAPOX), necrobiotic xanthogranuloma (NBX), and Erdheim–Chester disease (ECD). The association between orbital xanthogranuloma and other autoimmune diseases may be present. In a patient series, the co-occurrence of asthma, psoriasis, granuloma annulare, and idiopathic thrombocytopenic purpura was noted [59].
Epidemiology
Xanthogranulomatous disease of the orbit is rare, representing 5 % of all inflammatory orbital lesions and only 1.5 % of all orbital diseases in our series, making prospective evaluation or meta-analysis impossible. Usually, the onset is in middle age.
Etiology
According to the 2008 WHO classification [60], the disease should be classified as a dendritic cell disorder. Dendritic cells are derived from myeloid stem cells and the circulating peripheral blood monocyte pool. Dendritic cell disorders include Langerhans cell histiocytosis (LCH) (CD1a, S-100 positive) and non-LCH (CD1a -, CD68+, FXIIIa+), including juvenile xanthogranuloma, solitary reticulohistiocytoma, and ECD. In a retrospective case series, we have identified IgG4-positive plasma cells in 50 % of all orbital xanthogranulomas indicating an immunomodulatory mechanism that involves IgG4 production in many cases [61].
Localization
Patients classified as AOX, AAPOX, and NBX typically have preseptal and anterior orbital involvement. ECD tends to be diffuse and intraconal. Yellow skin lesions are found in all the syndromes [59].
Genetics
No genetic abnormalities have been reported except for ECD.
Erdheim–Chester Disease (ECD)
Erdheim–Chester disease (synonyms: Erdheim–Chester syndrome, polyostotic sclerosing histiocytosis). The disease involves an infiltration of lipid-laden macrophages, multinucleated giant cells, an inflammatory infiltrate of lymphocytes and histiocytes, and varying fibrosclerosis. It is the most devastating of the adult xanthogranulomas and is characterized by dense, progressive, recalcitrant fibrosclerosis of the orbit and internal organs, including the mediastinum; pericardium; pleural, perinephric, and retroperitoneal spaces; the bone marrow; and a generalized sclerosis of the long bones. A more complete description is given below in Sect. 12.7.11.2.
Adult-Onset Xanthogranuloma (AOX)
AOX is an isolated xanthogranulomatous lesion without systemic involvement and is associated with immune dysfunction: lymphoproliferative disease (benign/malignant) [59]. The extraocular muscles, adnexal tissue, and lacrimal gland may be affected by an infiltration of foamy histiocytes, Touton giant cells, lymphocytes, and varying degrees of fibrosis (Fig. 12.29). Adult-onset xanthogranuloma is the least aggressive of the xanthogranulomatous syndromes. It is strictly limited to the anterior orbital tissue, without any systemic findings, internal organ involvement, or immune dysfunction such as monoclonal gammopathy or lymphoproliferative disease.
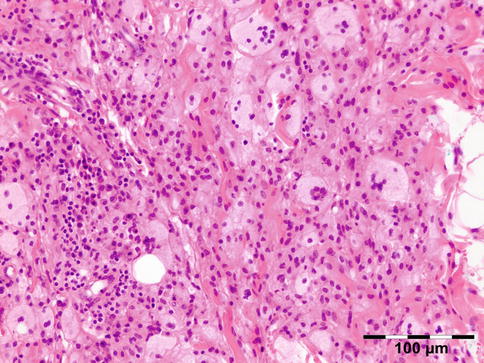
Fig. 12.29
Adult-onset xanthogranuloma (AOX): the tissue is infiltrated by foamy histiocytes, Touton giant cells, and lymphocytes with a low degree of fibrosis (HE 200×) (Dr. Robert M. Verdijk)
Adult-Onset Asthma with Periocular Xanthogranuloma (AAPOX)
AAPOX is a syndrome that was described by Jakobiec et al. [62]. It is rare (approximately 25 cases reported in the literature) and affects adults aged between 22 and 74 years, with a male to female ratio of 2:1. The association with lymphadenopathy suggests an underlying stimulation and proliferation of B-cell populations [59]. It affects the eyelids and orbit. Orbital involvement affects the anterior and preseptal compartments. Intraconal involvement has only been reported in one patient. AAPOX always presents with bilateral yellow or orange, indurated, and nonulcerated xanthomatous periocular masses. In addition to asthma, cases of AAPOX often have increased polyclonal IgG levels and lymphadenopathy. Histopathologic examination typically finds foamy histiocytes, Touton giant cells, and large lymphoid aggregates with classic germinal centers and no necrosis or fibrosis (Fig. 12.30). AAPOX is not a sight-threatening disease, but visual function can be impaired in the long term by eyelid occlusion or extraocular muscle infiltration.
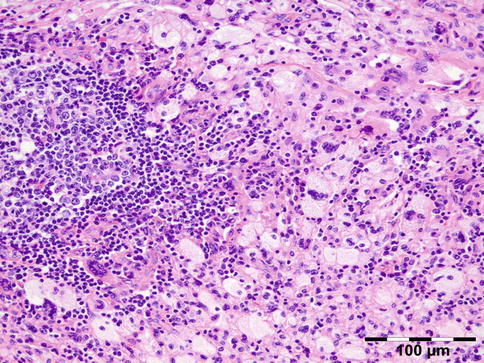
Fig. 12.30
Adult-onset asthma with periocular xanthogranuloma (AAPOX): like in all xanthogranulomas, there are foamy histiocytes and Touton giant cells. Note the large lymphoid aggregates with classic germinal centers (HE 200×) (Dr. Robert M. Verdijk)
Necrobiotic Xanthogranuloma
NBX is associated with immune dysfunction with monoclonal paraproteinemia (80 %), hypocomplementemia, cryoglobulinemia, multiple myeloma, and non-Hodgkin lymphoma (NHL) [62]. It is localized in the subcutaneous skin of the eyelids and anterior orbit but also occurs throughout the body. NBX affects adults (mean age 60 years). Although skin lesions are seen in all the syndromes, those in NBX have a strong propensity to ulcerate and fibrose. Internal organ involvement occurs in NBX but is often found only at autopsy. On histology there is a xanthogranulomatous inflammation with foamy histiocytes, Touton giant cells, and varying degrees of fibrosis. The presence of necrobiosis is required. Necrosis with palisading epithelioid histiocytes differentiates it from the other adult orbital xanthogranulomatous diseases (Fig. 12.31).
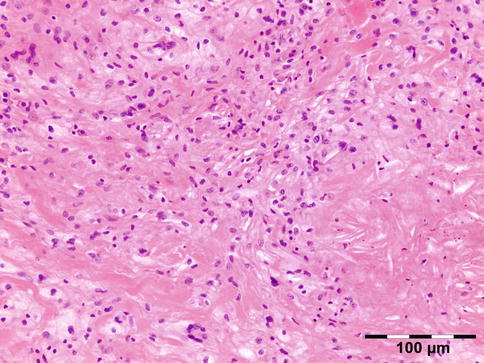
Fig. 12.31
Necrobiotic xanthogranuloma (NBX): in addition to a xanthogranulomatous inflammation with foamy histiocytes and Touton giant cells, there is necrosis surrounded by palisading epithelioid histiocytes. Fibrosis is another feature shown (HE 200×) (Dr. Robert M. Verdijk)
Prognosis
Time to development of associated malignancy ranges from 8 years before the skin lesions to 11 years after the skin lesions. Overall survival is 100 % at 10 years and 90 % at 15 years [63].
Juvenile Orbital Xanthogranuloma (JOX)
JOX, occurs only in children, patients are under 20 years of age (80–90 % under 2 years). It is localized in the orbit [64], and eyelids, conjunctiva, and iris may be involved. Patients usually present with yellow-red skin nodules that regress spontaneously. Iris infiltration can cause hyphema and glaucoma. Histopathology reveals an infiltrate of histiocytes with a finely vacuolated or xanthomatous cytoplasm, lymphocytes, and eosinophils. JOX pathology is notable for Touton giant cells. Immunohistochemistry shows positive staining for factor XIIIa, CD68, CD163, fascin, and CD14 and negative staining for CD1A, GFAP, desmin, keratin, CD99, and S-100. The differential diagnosis consists of xanthoma, other non-LCH, granulocytic sarcoma, or solitary fibrous histiocytoma. JOX has been described in patients suffering from NF1 or NF2. One case with a chromosomal aberration 46,XY,inv(13)(q21q33)c has been described [65]. Lesions usually regress or stabilize with time and have an excellent prognosis. Surgical excision is the mainstay treatment of choice in solitary lesions.
Wegener’s Granulomatosis (WG)
Definition
Small vessel vasculitis with autoimmune attack by ANCAs (antineutrophil cytoplasmic antibodies).
Synonyms
Granulomatosis with polyangiitis, ANCA-associated granulomatous vasculitis (professional bodies have advocated a more descriptive name since 2000 [66]; however this has not been generally adopted).
Epidemiology
WG represented 4 % of inflammatory lesions in our series. The incidence is ten cases per million per year. Ninety percent of the patients are white. While it mainly occurs in the middle-aged, it has been reported in much younger and older patients.
Etiology
It is presumed that the ANCAs are responsible for the inflammation in WG.
Localization
WG may present as an orbital mass without obvious upper respiratory or systemic features, but often is associated with ear nose and throat features and/or scleritis.
Clinical Features
C-ANCA testing is initially often negative but may become positive during late reactivation [67].
Macroscopy
Fibrotic, often hemorrhagic tissue fragments.
Histopathology
Orbital biopsies typically have features of mixed sclerosing, often angiocentric, inflammation, with fat disruption and small areas of necrosis, and focal microabscesses (Fig. 12.32). The findings of smudgy necrosis must not be regarded as a nonspecific finding (Fig. 12.33) [67]. Often there is no presence of fibrinoid necrosis of the vessels.
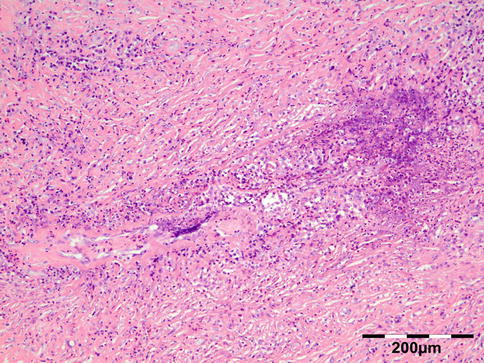
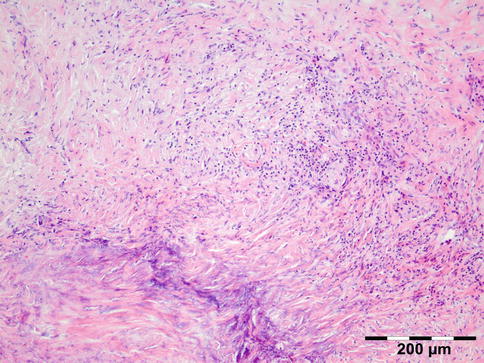
Fig. 12.32
Wegener’s granulomatosis: there is a mixed inflammatory cell population containing lymphocytes, histiocytes, eosinophilic, and notably neutrophilic polymorphonuclear cells in an angiocentric orientation and necrotic vasculitis. There is a severe sclerosis and areas of necrosis (HE 100×) (Dr. Robert M. Verdijk)
Fig. 12.33
Wegener’s granulomatosis: smudgy necrosis as depicted in the lower left of the photograph is not an aspecific finding in orbital inflammatory pseudotumors (HE 100×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Infectious granulomatous vasculitis, temporal arteritis, polyarteritis nodosa.
Histogenesis
Not applicable.
Genetics
HLA-DPB1 association with WG has been described; it is still the strongest and best replicated risk locus for this condition. Numerous other associations, including CTLA4, CD226, and copy number polymorphisms of FCGR3B, still need further investigation, before reliable conclusions can be drawn.
Prognosis and Predictive Factors
Twenty-five to forty percent of patients suffer from flare-ups, but a majority respond well to treatment. Relapses can be long and troublesome. Long-term complications are very common (86 %): mainly chronic renal failure, hearing loss, and deafness. Patients with the limited form of WG have a better prognosis because of the absence of kidney disease.
Vasculitis
Noninfectious vasculitides, apart from WG, comprise a large number of diseases. Many of these diseases can affect the orbit and lead to a clinical presentation, which mimics numerous other orbital processes. Orbital disease can be the initial presentation of a systemic vasculitis and early diagnosis can help prevent long-term, potentially fatal, consequences [68]. Panniculitis, also known as lupus erythematosus profundus involving orbital structures as the primary presenting symptom of SLE, has been described [69]. Churg–Strauss syndrome has been described in patients presenting with ocular manifestations and concurrent ear, nose, and throat symptoms and arthralgia or with positive ANCA and eosinophilia (Fig. 12.34) [70]. In rare cases orbital infarction may be associated with giant cell arteritis (Fig. 12.35) [71].
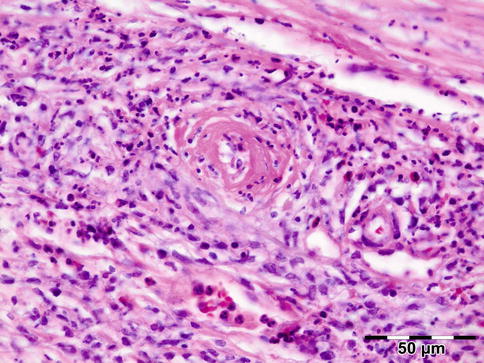
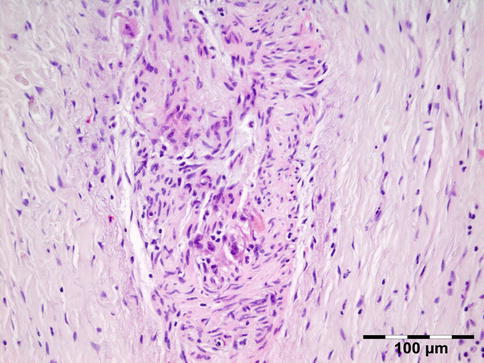
Fig. 12.34
Churg–Strauss syndrome: depicted is a necrotic vasculitis with a notable eosinophilic infiltrate and fibrosis. The histology is comparable to Wegener’s granulomatosis. This patient had peripheral blood eosinophilia in addition to positive ANCA (HE 400×) (Dr. Robert M. Verdijk)
Fig. 12.35
Giant cell arteritis: thick-walled muscular artery infiltrated by lymphocytes, histiocytes, and multinucleated giant cells. In the fibrotic surrounding stroma admixed eosinophils are present (HE 400×) (Dr. Robert M. Verdijk)
Sarcoidosis
Sarcoidosis is a multisystem disorder characterized histopathologically by noncaseating granulomas. It affects both sexes equally, and although it can present at any age, it usually develops before 50 years of age. In the United States, the annual incidence of sarcoidosis is estimated to be 36 in 100,000 for black persons and 11 in 100,000 for white persons, with 3.8-fold greater risk of developing sarcoidosis in black persons. Sarcoidosis most commonly involves the lungs and the mediastinal lymph nodes (86–92 %), the eye and ocular adnexa (10–50 %), the peripheral lymph nodes (33 %), the skin (10–40 %), the central nervous system (10 %), and the heart (5 %). Sarcoidosis can affect any part of the eye, orbit, and adnexal structures. Orbital and adnexal involvement includes the lacrimal gland in 63 % (19/30), the eyelid in 17 % (5/30), the orbit in 13 % (4/30), and the lacrimal sac in 7 % (2/30) [72]. Sarcoidosis represented 9 % of inflammatory lesions in our series and 2.5 % of all orbital lesions. Orbital and adnexal sarcoidosis usually presents as enlargement of the lacrimal gland(s) but also can present as a diffuse, solid mass with irregular infiltration of orbital structures, extraocular muscles, or lacrimal sac. The most common signs are those of inflammation: erythema (70 %) and edema of the eyelids (36 %). Thirty-seven percent of patients with periorbital sarcoidosis (11/30) have known systemic disease [72]. Of 63 % (19/30) of cases with only orbital disease, systemic disease initially was found in 27 % (8/30), subsequently in 7 % (2/30), and never in 30 % (9/30) [72]. On computed tomography scans, the lesions were well circumscribed in 85 % (25/30) and diffuse in 15 % (5/30) [72]. The lesion consists of generally non-necrotizing granulomas (Fig. 12.36). Management includes systemic steroids, excision, and observation. Periocular steroid injections have been used for localized orbital disease with good results.
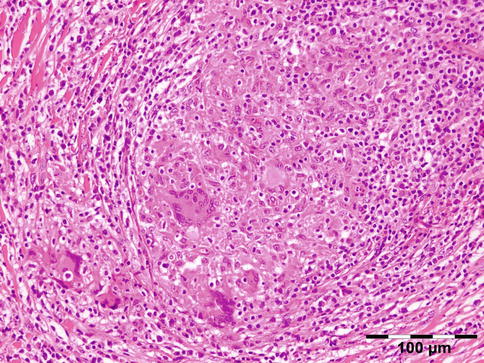
Fig. 12.36
Sarcoidosis: a central non-necrotizing granuloma consisting of histiocytes and multinucleated giant cells is surrounded by lymphocytes and plasma cells. There is a severe fibrosis of the stroma (HE 200×) (Dr. Robert M. Verdijk)
12.5 Injuries
12.5.1 Traumatic Neuroma
Definition
A proliferation of Schwann cells and connective tissue on the proximal end of a severed nerve.
Epidemiology
Unknown; may occur more often postsurgery without clinical symptoms. Traumatic neuroma represented less than 1 % of all orbital lesions in our series and 19 % of all neurogenic tumors.
Etiology
New axons emerge and tend to follow the Schwann cells.
Localization
At the site of previous trauma or surgical procedure.
Clinical Features
The mass may assume tumorlike proportions. Simple resection is the proper treatment option [73].
Macroscopy
Fibrotic nerve tissue.
Histopathology
The erratically ensheathed axons are embedded in scar tissue and course in all directions (Fig. 12.37).
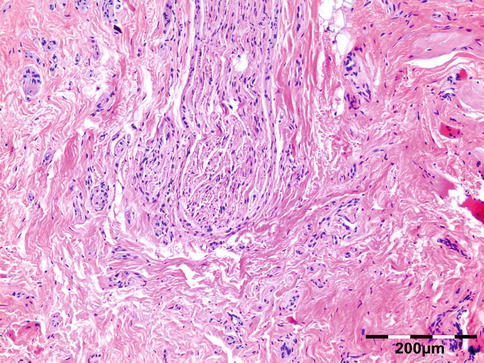
Fig. 12.37
Traumatic neuroma: centrally a nerve fiber is shown in a fibrotic stroma surrounded by erratically ensheathed axons that course in all directions (HE 100×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Neurofibroma, perineurioma.
Histogenesis
The orbital nerves are derived from the cranial nerves.
Genetics
Not applicable.
Prognosis and Predictive Factors
Recurrence of traumatic neuroma may occur after resection.
12.6 Degenerations
12.6.1 Fat/Lacrimal Gland Prolapse
Definition
Fat/lacrimal gland prolapse is an acquired lesion, characterized by a herniation of intraconal fat due to weakness of the Tenon capsule by the aging process, trauma, or surgery.
Epidemiology
In adults it is associated with elderly adipose patients. Fat and lacrimal gland prolapse represented 2 % of all lesions in our series and 35 % of all degenerative lesions.
Etiology
Localization
Fat prolapse usually occurs in the lateral canthal area beneath the temporal or superotemporal bulbar conjunctiva.
Clinical Features
Patients with subconjunctival fat prolapse present with a fatty mass in the lateral canthal area either unilaterally or bilaterally.
Macroscopy
Normal fatty tissue is observed.
Histopathology
Normal fatty tissue is observed.
Differential Diagnosis
Fat prolapse and dermolipoma are two distinct entities of the orbit that are often confused clinically. The distinction with extremely rare lipoma of the orbit cannot be made on a histopathologic basis.
Histogenesis
Orbital fat is of mesenchymal origin as is the orbital septum.
Genetics
No genetic defects have been described.
Prognosis and Predictive Factors
Surgical removal through a perilimbal conjunctival incision can easily be done if the lesion is cosmetically unacceptable or causes discomfort.
12.6.2 Amyloidosis
Definition
Amyloidosis is a disorder of protein metabolism characterized by the extracellular deposition of abnormal protein fibrils.
Epidemiology
Orbital amyloidosis is extremely rare regardless of the etiology and represented only two cases (far less than 1 %) in our series.
Etiology
A classification system divides amyloidoses into three categories: (1) local amyloidosis, (2) hereditary systemic amyloidosis, and (3) acquired systemic amyloidosis [75]. Acquired systemic amyloidoses can be further subdivided into systemic AL amyloidosis (previously known as primary amyloidosis), reactive systemic AA amyloidosis (previously known as secondary amyloidosis), Aβ2M amyloidosis (dialysis-associated), and ATTR amyloidosis (senile amyloidosis). Systemic AL amyloidosis is almost always associated with a plasma cell dyscrasia, with multiple myeloma accounting for 20 % of these cases. Controversy exists in the literature as to whether orbital amyloidosis is more frequently due to primary localized disease or secondary to systemic amyloidosis.
Localization
It may be localized [76] or distributed throughout the body, causing organ damage and serious morbidity.
Clinical Features
The implication of missing a potentially life-threatening disease necessitates that a systemic investigation be completed before the diagnosis of primary localized orbital amyloidosis is made [77].
Macroscopy
The biopsy consists of fibroadipose tissue with a flesh-colored to yellow waxy appearance.
Histopathology
The tissues show interstitial deposits of eosinophilic amorphous hyaline material. Complete diagnosis of amyloid may be carried out in two steps on tissue sections. Congo red stain reveals the hyaline stromal deposits (Fig. 12.38) to demonstrate apple-green birefringence under polarized light (Fig. 12.39). The second diagnostic step is the identification of the amyloid protein. Immunohistochemistry using a panel of antibodies directed against kappa or lambda light chain, AA protein, ATTR protein, and Abeta2M may be used.
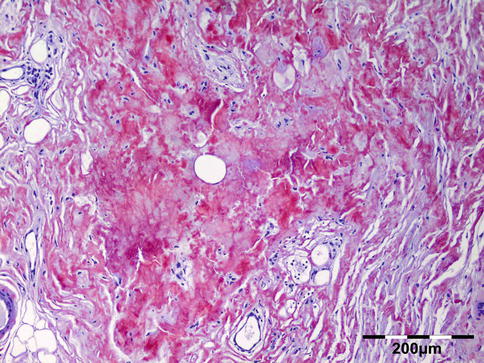
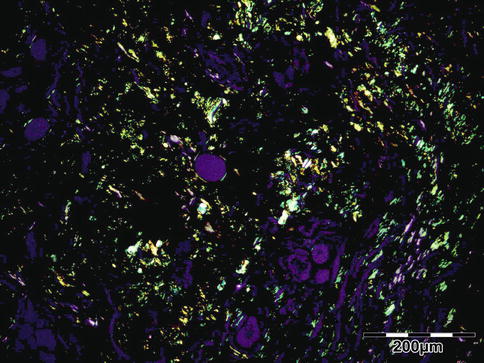
Fig. 12.38
Amyloidosis: the fat tissue shows interstitial deposits of eosinophilic amorphous hyaline material (Congo red 100×) (Dr. Robert M. Verdijk)
Fig. 12.39
Amyloidosis: apple-green birefringence can be demonstrated under polarized light (Congo red, polarized light 100×) (Dr. Robert M. Verdijk)
Genetics
ATTR amyloid is associated with hereditary syndromes including familial amyloid polyneuropathies and familial amyloid cardiomyopathies and can be diagnosed by molecular genetic analysis [78].
Prognosis and Predictive Factors
Each of the chemical amyloid types has a different prognosis and requires a different therapy.
12.7 Neoplasms
Neoplasms may vary from benign to high-grade malignant. In soft tissue sarcomas the modified three-tiered FNCLCC system is based on a score obtained by evaluating three parameters: tumor differentiation, mitotic rate, and amount of tumor necrosis. A score is attributed independently to each parameter and the grade is obtained by adding the three attributed scores. Tumor differentiation: Score 1: sarcomas closely resembling normal adult mesenchymal tissue (e.g., low-grade leiomyosarcoma). Score 2: sarcomas for which histologic typing is certain (e.g., myxoid liposarcoma). Score 3: embryonal and undifferentiated sarcomas, sarcomas of doubtful type, synovial sarcomas, osteosarcomas, and PNET. Mitotic count: Score 1: 0–9 mitoses per 10 HPF. Score 2: 10–19 mitoses per 10 HPF. Score 3: >20 mitoses per 10 HPF. Tumor necrosis: Score 0: no necrosis. Score 1: <50 % tumor necrosis. Score 2: >50 % tumor necrosis. Histologic grade: Grade 1: total score 2, 3. Grade 2: total score 4, 5. Grade 3: total score 6, 7, 8.
12.7.1 Fibroblastic/Myofibroblastic/Fibrohistiocytic Tumors
12.7.1.1 Nodular Fasciitis
Synonyms
The original designation of subcutaneous pseudosarcomatous fibromatosis should be avoided.
Epidemiology
Nodular fasciitis occurs in all age groups but more often in young adults. There is no sex predilection for nodular fasciitis or intravascular fasciitis, but cranial fasciitis is more frequent in boys. We only encountered one case in our series.
Etiology
Unknown.
Localization
Nodular fasciitis is typically located anteriorly in the periorbital area and is well circumscribed. Nodules most commonly develop in the subcutaneous or superficial fascia of the extremities. Nodular fasciitis is rarely observed in the head and neck region of adults; however, these locations are more common in infants and children [80]. In children it tends to occur in the anterior periorbital tissues, but it has also been reported to occur in the orbit where it can simulate a dermoid cyst [81]. Other cases involved the Tenon capsule [82, 83].
Clinical Features
Mostly these are solitary masses that grow rapidly in a course of several weeks. In about 50 % of the cases the swelling is painful.
Macroscopy
Nodular fasciitis may appear circumscribed or infiltrative but has no capsule or pseudocapsule. The cut surface varies from myxoid to fibrous, and occasionally there is central cystic change.
Histopathology
Classically there is the proliferation of spindle cell fibroblasts which are frequently arranged in parallel bundles extending in all directions resembling cells in tissue culture (Fig. 12.40). Mitoses may be numerous, but the spindle cell nuclei are never hyperchromatic and atypical mitoses are virtually never seen [84, 85]. The lesion may be highly cellular, but typically it is at least partly loose appearing and myxoid. There is normally little collagen. Extravasated red cells, chronic inflammatory cells, and multinucleated osteoclast-like giant cells are frequently identified. The lesional border is typically, at least focally, infiltrative. A vascular component may consist of capillary spaces or endothelial cells lined up in columns. Stains for SMA and MSA are usually positive, consistent with myofibroblastic differentiation but this does not distinguish nodular fasciitis from many other mesenchymal proliferations. CD68 staining is present in the osteoclast-like giant cells and occasionally in spindle cells. Keratin and S-100 protein are typically negative.
Fig. 12.40
Nodular fasciitis: spindle cell fibroblasts are arranged in parallel bundles extending in all directions resembling cells in tissue culture loose appearing and myxoid. There are extravasated red cells and few chronic inflammatory cells (HE 100×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Myxoma, fibrous histiocytoma, fibromatosis, inflammatory myofibroblastic tumor (IMT), sarcoma (fibrosarcoma, myxofibrosarcoma). The lesion is myofibroblastic in nature.
Genetics
Normal diploid DNA.
Prognosis and Predictive Factors
There is no evidence of aggressive clinical behavior, recurrence, or metastasis.
12.7.1.2 Myofibroma/Myofibromatosis
Definition
Myofibroma and myofibromatosis are terms used to denote the solitary (myofibroma) or multicentric (myofibromatosis) occurrence of benign neoplasms composed of myoid cells arranged around thin-walled blood vessels [79].
Synonyms
Congenital fibrosarcoma, congenital generalized fibromatosis, infantile myofibromatosis.
Epidemiology
Although rare, with only two cases in our series, myofibroma is the most common fibrous tumor of childhood. Solitary cases are more common in males.
Etiology
Infantile myofibromatosis was first described by Stout in 1954 as “congenital generalized fibromatosis” and was renamed as infantile myofibromatosis by Chung and Enzinger in 1981 after recognition of the myofibroblastic nature of the lesion (referenced in [86]). Myofibroma(tosis) forms a morphological continuum with myopericytoma and so-called infantile hemangiopericytoma and infantile myofibromatosis.
Localization
Clinical Features
Solitary myofibroma is characterized by the presence of one nodule in the skin, muscle, bone, or subcutaneous tissue. The multicentric type can be divided into two subtypes: multicentric lesions with or without visceral involvement. The tumors are benign, nonencapsulated, and locally infiltrating and generally follow a benign course. Isolated orbital infantile myofibromas are rare tumors in the head and neck. The mass-like clinical presentation and variable histologic features result in frequent misdiagnosis and potentially inappropriate clinical management. Twenty-four patients with orbital infantile myofibroma or myofibromatosis were reviewed [87] aged newborn to 10 years (mean, 34.8 months), who presented with a painless mass in the infra- or supraorbital regions, usually increasing in size and associated with exophthalmos. The tumors were twice as frequent on the left as on the right. Patients experienced symptoms for an average of 2.7 months before clinical presentation. The tumors involved the bone or the soft tissues of the orbit, some with extension into the nasal or oral cavity.
Macroscopy
The mean tumor size is 3.0 cm. Grossly, tumors are well circumscribed, firm to rubbery, homogeneous, and white gray. Myofibromas have a firm, fibrous cut surface and are grayish white, light tan to brown, or purplish in color. They often have central yellow/necrotic areas and/or cystic spaces filled with caseous-like material or hemorrhage.
Histopathology
Histologically characterized by a (multi)nodular mitotic active proliferation of spindly shaped myofibroblasts without significant atypia [90]. Myofibroblasts are spindle shaped with pale pink cytoplasm and have elongated, tapering nuclei with a vesicular chromatin pattern and one or two small nucleoli (Fig. 12.41). Myoid whorls or nodules often hyalinize, with a pseudochondroid appearance. Within the center of the nodules, there are less well-differentiated, rounded, polygonal, or spindle-shaped cells, with slightly larger, hyperchromatic nuclei. These cells have relatively scant cytoplasm and are arranged around thin-walled, irregularly branching, hemangiopericytoma-like blood vessels. Both the myofibroblastic and more primitive components are positive for vimentin and smooth muscle actin, while the myofibroblastic component is more strongly positive for pan-actin HHF-35. Both components are negative for S-100 protein, epithelial membrane antigen, and keratin.
Fig. 12.41
Myofibromatosis: spindle-shaped myofibroblasts with pale pink cytoplasm and elongated, tapering nuclei with a vesicular chromatin pattern can be seen in a fascicular pattern and formation of myoid whorls (HE 200×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Nodular fasciitis, neurofibroma, infantile fibromatosis, fibrous histiocytoma, juvenile hyaline fibromatosis, fibrous hamartoma of infancy, fibromatosis colli, leiomyoma, infantile hemangiopericytoma, infantile fibrosarcoma, Ewing sarcoma/primitive neuroectodermal tumor, and lymphoma.
Histogenesis
The lesion is myofibroblastic in nature.
Genetics
Sporadic cases with del(6)(q12;q15), monosomy 9q, and trisomy 16q have been reported.
Prognosis and Predictive Factors
The extent of the disease is the most important prognostic factor. Solitary lesions often resolve after (incomplete) resection. Recurrences, however may develop. Complete excision with close follow-up is the preferred treatment [91].
12.7.1.3 Juvenile Hyaline Fibromatosis
Juvenile hyaline fibromatosis has rarely been described as an eyelid tumor scalloping the superior orbital osseous rim [92]. Fibrous hamartoma of infancy and fibromatosis colli have not been described in the orbit.
12.7.1.4 Infantile Hemangiopericytoma
Infantile hemangiopericytoma occurs rarely in the orbit [93]. It is currently considered that a broad spectrum of benign infantile myofibroblastic lesions exists containing an immature-appearing cellular component with a distinctive, hemangiopericytoma-like vascular pattern. Infantile myofibromatosis and so-called infantile hemangiopericytoma almost certainly represent different stages of maturation of the same (single) entity [94].
12.7.1.5 Inflammatory Myofibroblastic Tumor (IMT)
Definition
Inflammatory myofibroblastic tumor (IMT) is a lesion composed of myofibroblastic spindle cells accompanied by an inflammatory infiltrate of plasma cells, lymphocytes, and eosinophils [79].
Epidemiology
Rare; we only encountered one case in our series.
Etiology
Inflammatory myofibroblastic tumor (IMT) is considered to be a neoplastic counterpart of idiopathic inflammatory pseudotumor (IIP).
Localization
Clinical Features
Painful exophthalmos of the eye.
Macroscopy
Lobular, multinodular, or bosselated with a hard rubbery white, gray, yellow, or red cut surface.
Histopathology
Histopathology is characterized by a predominantly myofibroblastic lesion. Plump or spindled myofibroblasts may be in an edematous myxoid background with abundant blood vessels and an infiltrate of plasma cells, lymphocytes, and eosinophils (Fig. 12.42) to resemble granulation tissue, nodular fasciitis, or other reactive processes. Another pattern is characterized by a compact fascicular spindle cell proliferation with variable myxoid and collagenized regions and a distinctive inflammatory infiltrate with diffuse inflammation, small aggregates of plasma cells, or lymphoid nodules resembling fibromatosis, fibrous histiocytoma, or a smooth muscle neoplasm. Ganglion cell-like myofibroblasts with vesicular nuclei, eosinophilic nucleoli, and abundant amphophilic cytoplasm can be seen. Certain lesions resemble a scar or desmoid-type fibromatosis, with platelike collagen, lower cellularity, and relatively sparse inflammation with plasma cells and eosinophils. Highly atypical polygonal cells with oval vesicular nuclei, prominent nucleoli, and variable mitoses, including atypical forms, are seen in rare IMTs which undergo histologic malignant transformation. Subsets of IMT show aberrant expression of anaplastic lymphoma kinase (ALK), and immunohistochemical expression of ALK is noted in 68 % of the cases (Fig. 12.43) [99]. However, ALK positivity is not specific for IMT. Strong diffuse cytoplasmic reactivity for vimentin is present in virtually all IMT. Reactivity for smooth muscle actin and muscle-specific actin varies from a focal to a diffuse pattern in the spindle cell cytoplasm, and desmin is identified in many cases. Focal cytokeratin immunoreactivity is seen in about one-third of cases. Myogenin, myoglobin, and S-100 protein are negative. TP53 immunoreactivity is rare and has been reported in association with recurrence and malignant transformation.
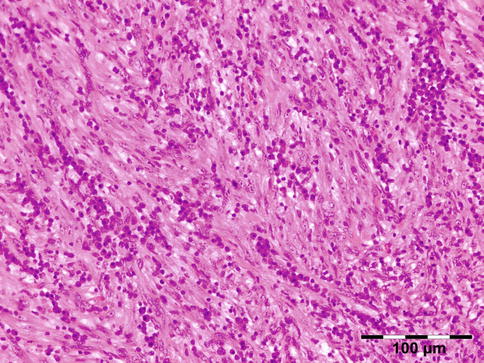
Fig. 12.42
Inflammatory myofibroblastic tumor (IMT): the lesion is composed of a compact fascicular pattern of plump myofibroblasts with an infiltrate of mainly plasma cells and admixed lymphocytes (HE 200×) (Dr. Robert M. Verdijk)
Fig. 12.43
IMT: the myofibroblasts stain positive for ALK1 (ALK1 IH 200×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Inflammatory malignant fibrous histiocytoma, Hodgkin lymphoma, xanthogranulomatous inflammation secondary to Erdheim–Chester disease, pseudotumor resulting from (atypical) mycobacterial infection, inflammatory fibrosarcoma.
Histogenesis
The lesion is myofibroblastic in nature.
Genetics
IMTs are heterogeneous genetically. ALK rearrangements occur mainly in children and young adults and are detectable by fluorescence in situ hybridization. The cytogenetic rearrangements activate the ALK receptor tyrosine kinase gene in chromosome band 2p23. IMT is therefore considered to be a clonal neoplastic lesion. By contrast, such rearrangements are uncommon in IMT diagnosed in adults beyond 40 years old.
Prognosis and Predictive Factors
Cellularity or mitotic counts are not prognostic markers. Cytological atypia, ganglion cell-like cells, and p53 expression may identify tumors that are more likely to pursue an aggressive course.
12.7.1.6 Solitary Fibrous Tumor/Hemangiopericytoma/Giant Cell Angiofibroma
Definition
Tumors of benign to uncertain behavior, CD34-positive, collagen-rich, specialized fibroblastic lesions, which have overlapping or histologically identical features [79].
Epidemiology
Rare; we only encountered one case of hemangiopericytoma in our series. These are tumors of adults with an equal sex distribution.
Etiology
Recently, a large series of these collagen-rich fibroblastic tumors of the orbit has been explored. Forty-one fibroblastic orbital tumors, originally diagnosed as hemangiopericytoma (n = 16), fibrous histiocytomas (n = 9), mixed tumors (hemangiopericytoma/fibrous histiocytoma) (n = 14), and giant cell angiofibromas of orbit (n = 2), were evaluated. Upon histologic review with the addition of CD34 immunostaining, all cases were reclassified as solitary fibrous tumor [100].
Localization
Hemangiopericytoma and solitary fibrous tumors with the addition of giant cell angiofibromas are uncommon neoplasms found in deep soft tissue on many locations, including the orbit.
Clinical Features
Patients may present with an usually painless orbital mass and proptosis. Duration of symptoms may range from 3 to 96 months [100]. Many (41 %) patients present with an orbital mass, 20 % with proptosis, 5 % with painful mass, and 5 % with painless mass.
Macroscopy
Most SFTs present as well-circumscribed, often partially encapsulated, masses, measuring from 0.4 to 5.0 cm [100]. On section, they frequently have a multinodular, whitish, and firm appearance; myxoid and hemorrhagic changes are occasionally observed. Tumor necrosis and infiltrative margins (about 10 % of cases) are mostly observed in locally aggressive or malignant tumors.
Histopathology
Typical SFTs show a patternless architecture characterized by a combination of alternating hypocellular and hypercellular areas separated from each other by thick bands of hyalinized, somewhat keloidal, collagen and branching staghorn-like vessels (Fig. 12.44). The non-atypical, round to spindle-shaped tumor cells have little cytoplasm with indistinct borders and dispersed chromatin within vesicular nuclei. Myxoid change, areas of fibrosis, and interstitial mast cells are commonly observed. Mitoses are generally scarce, rarely exceeding 3 mitoses per 10 high-power fields. SFTs may contain mature adipocytes and/or giant multinucleated stromal cells which used to be restricted to so-called lipomatous hemangiopericytoma and giant cell angiofibroma. Malignant SFTs are usually hypercellular lesions, showing at least focally moderate to marked cytological atypia, tumor necrosis, numerous mitoses (>4 mitoses per 10 high-power fields), and/or infiltrative margins. Rare cases show abrupt transition from conventional benign-appearing SFT to high-grade sarcoma, likely representing a form of dedifferentiation. However, overall outcome for malignant solitary fibrous tumors of the eye should be further explored [100]. Tumor cells in SFT are characteristically immunoreactive for CD34. Twenty to thirty-five percent of them are also variably positive for epithelial membrane antigen, BCL-2, and smooth muscle actin. Focal and limited reactivity for S-100 protein, cytokeratins, and/or desmin has also occasionally been reported.
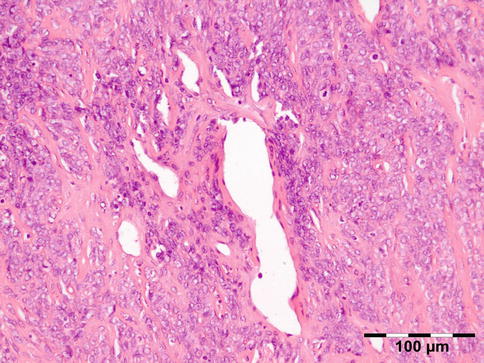
Fig. 12.44
SFT/Hemangiopericytoma: a hypercellular area is shown with round to spindle-shaped tumor cells that have little cytoplasm with indistinct borders and dispersed chromatin within vesicular nuclei. There is a collagenous background with branching vessels. Mitoses can be observed and should not exceed 3 mitoses per 10 high-power fields (HE 200×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Fibrous histiocytoma, mesenchymal chondrosarcoma, glomangioma.
Histogenesis
The lesion is fibroblastic in nature.
Genetics
SFTs are cytogenetically heterogeneous, the most common finding being rearrangements of the long arm of chromosome 12.
Prognosis and Predictive Factors
Local recurrences of SFT are possible and usually follow an incomplete initial excision. Recurrent tumors in the orbit have shown the tendency to infiltrate the surrounding tissues and the bone, rendering complete secondary excision more difficult [101].
12.7.1.7 Fibrous Histiocytoma
Epidemiology
Fibrous histiocytoma was in the past considered to be the most common primary mesenchymal orbital tumor of adults. In our series they represented only 6.5 % of all bone and soft tissue tumors and less than 1 % of all orbital lesions.
Etiology
Unknown.
Localization
The upper and nasal portions of the orbit are the most common sites of involvement.
Clinical Features
The tumors occur at a median age of 43 years. The most common signs and symptoms are proptosis (60 %), mass (46 %), and decreased vision (25 %).
Macroscopy
Usually the tumor consists of a lobulated circumscribed pseudo-encapsulated mass of variable size made up of rubbery-to-firm grayish-white, sometimes yellowish-tan, tissue. Occasionally, cystic or hemorrhagic reddish-brown areas may be noted, and in a few tumors, areas with a mucoid appearance are present. The size of the tumors range from 1 to 15 cm in greatest diameter. In contrast, malignant fibrous histiocytomas (MFH) usually display infiltrating margins and generally are larger than the benign lesions.
Histopathology
Deep fibrous histiocytomas usually show a prominent storiform pattern, sometimes combined with hemangiopericytoma-like areas. Contrary to conventional cutaneous lesions, most lesions show monomorphism and usually lack secondary elements such as foamy cells and giant cells but usually show scattered lymphocytes. Thus, they more closely resemble the cellular variant of cutaneous fibrous histiocytoma. The tumor cells are cytologically bland and generally spindle shaped with elongated or plump vesicular nuclei and eosinophilic, ill-defined cytoplasm. In the benign lesions, there is no nuclear pleomorphism or hyperchromasia, and mitoses, although commonly present, are usually less than 5 per 10 high-power fields. The stroma may show myxoid change or hyalinization and rarely osteoclast-like giant cells or metaplastic ossification. Small foci of necrosis may be present. Recurrent fibrous histiocytomas display infiltrating margins and areas of hypercellularity, but no significant nuclear pleomorphism or cellular atypia should be observed [102]. Malignant fibrous histiocytoma (MFH) show infiltrating edges as well as one or more histologic criteria of malignancy, including nuclear pleomorphism, bizarre multinucleated tumor giant cells, and areas of necrosis (Fig. 12.45) [102]. Immunohistochemistry shows similar results as in cutaneous lesions with negativity for epithelial markers, desmin, and S-100 protein. Alpha-smooth muscle actin may be positive in some parts of the lesion. CD34 is negative, and if positive, solitary fibrous tumor should be considered.
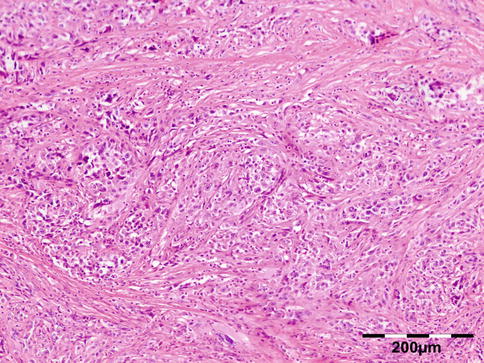
Fig. 12.45
Malignant fibrous histiocytoma (MFH): spindle to epithelioid cells with nuclear pleomorphism, bizarre multinucleated tumor giant cells, and atypical mitosis can be seen in a fibrotic stroma (HE 100×) (Dr. Robert M. Verdijk)
Differential Diagnosis
In a recent study, cases previously designated as fibrous histiocytoma showed overlapping morphologic and immunohistochemical features with giant cell angiofibroma, orbital hemangiopericytoma, and SFT and have been reclassified as SFT [100].
Histogenesis
The origin of the neoplasm is probably a primitive mesenchymal cell.
Genetics
The genetic aspects of benign fibrous histiocytomas and MFH are difficult to evaluate because of the shifting diagnostic criteria used throughout the years.
Prognosis and Predictive Factors
Based on the histopathologic features, the tumors have been classified in three groups: benign, locally aggressive, and malignant [102].
12.7.1.8 Myxoma and Angiomyxoma
Myxomas are rare, benign neoplasms of mesenchymal origin that usually develop in soft tissues. Myxomas of the orbit are rare. Median age at presentation is 56 years (range, 4–69). Two angiomyxomas occurred in children ages 4 and 7 years whose tumors were locally aggressive and recurred. Pathologically, the tumors are poorly circumscribed. Histopathology showes them to be hypocellular, containing stellate and spindled cells in an abundant, loose, myxoid stroma rich in hyaluronic acid. Small blood vessels are rare in myxomas but abundant in angiomyxomas (Fig. 12.46). Tumor cells are frequently immunoreactive for vimentin, CD34, and factor XIIIa. Vascularity and bone involvement appear to be important prognostic features for recurrence. Complete resection with a margin of healthy tissue appears to be the treatment of choice [103]. The differential diagnosis is with other neoplasms that may contain myxomatous areas. Multiple myxomas and angiomyxomas may be associated with Carney’s complex of multiple myxomas, blotchy skin pigmentation, schwannomas, and endocrine disturbances.
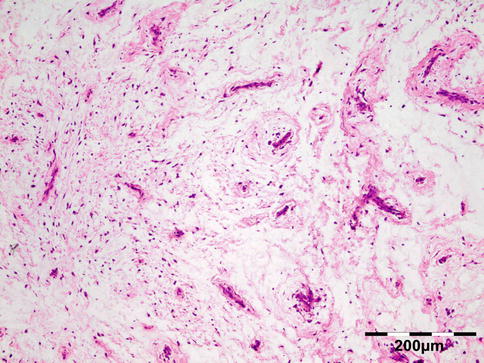
Fig. 12.46
Angiomyxoma: a hypocellular lesion composed of stellate and spindled cells in an abundant, loose, myxoid stroma rich in hyaluronic acid is observed. Abundant blood vessels are typical of angiomyxoma (HE 100×) (Dr. Ian W. McLean; this case was presented at the combined Verhoeff-EOPS meeting, Houston, 1996, case 96–14)
12.7.1.9 Myxofibrosarcoma
Definition
Myxofibrosarcoma (MFS), formerly known as myxoid variant of malignant fibrous histiocytoma (MFH), is a malignant mesenchymal tumor that comprises a spectrum of malignant fibroblastic lesions with variably myxoid stroma and pleomorphism and with a distinctively curvilinear vascular pattern.
Epidemiology
Rare.
Etiology
Unknown.
Localization
The most common locations of MFS are the limb and limb girdle followed by the head and neck, retroperitoneum, and mediastinum. There have been two cases described of MFS occurring in the orbit [104].
Clinical Features
MFS occurs in older adults.
Macroscopy
Superficial tumors typically consist of multiple, variably gelatinous or firmer nodules, whereas deep-seated neoplasms often form a single mass with an infiltrative margin. In high-grade lesions, areas of tumor necrosis are often found.
Histopathology
The histopathology is characterized by a hypocellular neoplasm composed of only few, non-cohesive, plump spindle or stellate tumor cells with ill-defined, slightly eosinophilic cytoplasm and atypical, enlarged, hyperchromatic nuclei in a myxoid stroma composed of hyaluronic acid. A characteristic finding is the presence of prominent elongated, curvilinear, thin-walled blood vessels with a perivascular condensation of tumor cells and/or inflammatory cells (mainly lymphocytes and plasma cells). Frequently, so-called pseudolipoblasts (vacuolated neoplastic fibroblastic cells with cytoplasmic acid mucin) are noted. Myxofibrosarcoma shows a broad spectrum of cellularity, pleomorphism, and proliferative activity. High-grade neoplasms are composed in large part of solid sheets and cellular fascicles of spindled and pleomorphic tumor cells with numerous, often atypical, mitoses, areas of hemorrhage, and necrosis. Bizarre, multinucleated giant cells with abundant eosinophilic cytoplasm (resembling myoid cells) and irregular-shaped nuclei are noted. Tumor cells stain positively for vimentin, and in a minority of cases, some spindled or larger eosinophilic tumor cells express muscle-specific actin and/or alpha-smooth muscle actin, suggestive of focal myofibroblastic differentiation; desmin, CD68, Mac 387, and FXIIIa are negative.
Differential Diagnosis
Myxoma, fibrosarcoma.
Histogenesis
The lesion is (myo)fibroblastic in nature.
Genetics
In general, the karyotypes tend to be highly complex, with extensive intratumoral heterogeneity.
Prognosis and Predictive Factors
The clinical behavior appears to be closely related to tumor grade with increased risk for local recurrence according to higher grades.
12.7.1.10 Infantile Fibrosarcoma (IF)
Infantile fibrosarcoma (IF) is a rare soft tissue sarcoma that presents either at birth or in the first year of life. IF has been once described as metastasis in the orbit [105].
12.7.2 Adipocytic Tumors
12.7.2.1 Lipoma
Epidemiology
Lipoma is the most common soft tissue neoplasm in adults but is extremely rare to occur in the orbit. We have no example in our series. A few spindle cell lipomas of the orbit have been reported [106, 107]. Our series also contained one spindle cell lipoma. Pleomorphic lipoma has been described in the orbit [108]. Intramuscular lipoma was only once described in the orbit [109].
Etiology and Localization
Diagnosed orbital lipomas in fact are often orbital fat prolapse.
Clinical Features
Are of an orbital mass.
Macroscopy
Lipomas should be well circumscribed and have a slightly less yellow tan, when compared to normal orbital fat. Different types are basically similar in appearance.
Histopathology
Conventional lipoma is composed of lobules of mature adipocytes. The cells are identical to the surrounding adipose tissue except for slight variation in the size and shape of the cells in lipomas. Lipomas can occasionally have areas of abundant fibrous tissue (fibrolipoma) [110]. Spindle cell lipoma is composed of bland mitotically inactive spindled cells arranged in parallel bands between the fat cells and associated with thick ropelike collagen bundles (Fig. 12.47). Pleomorphic lipoma is characterized by small spindled and rounded hyperchromatic cells and multinucleated giant cells with radially arranged nuclei. Immunohistochemistry may aid in the diagnosis of spindle cell and pleomorphic lipomas. Mature adipocytes stain positive for S-100 protein, vimentin, and CD34 (Fig. 12.48). The spindle cells in both spindle cell and pleomorphic lipomas on the other hand are strongly positive for CD34 and may rarely be positive for S-100 protein. Few cases of lipomatous hemangiopericytoma (adipocytic variant of solitary fibrous tumor) of the orbit have been described [111].
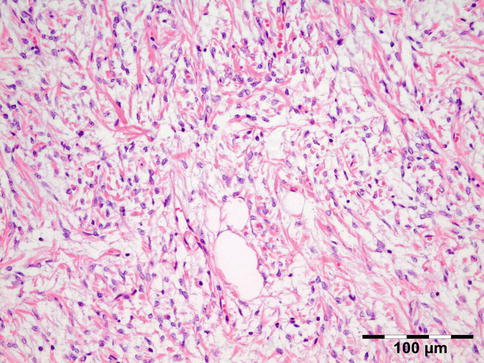
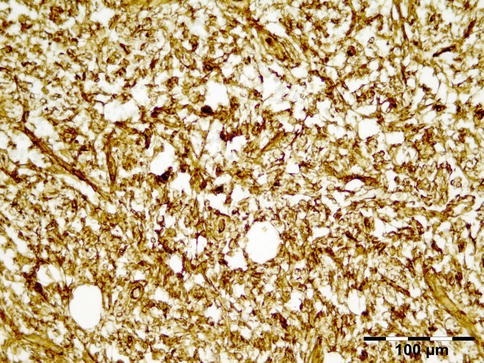
Fig. 12.47
Spindle cell lipoma: the lesion is composed of bland spindled cells arranged in parallel bands with occasional adipocytes. The thick ropelike collagen bundles are typical of the lesion. The molecular studies showed a typical polysomy signal with the MDM2 FISH probe (HE 200×) (Dr. Robert M. Verdijk)
Fig. 12.48
Spindle cell lipoma: the lesional cells stain positive for CD34 (CD34 IH 200×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Many reported cases have been simple orbital fat or fat prolapse.
Histogenesis
The adipocytes are of mesodermal origin.
Genetics
Chromosome aberrations have been found in 55–75 % of lipomas. The pattern of cytogenetic aberrations is heterogeneous. Chromosome 12 polysomy was noted in 89 % of spindle cell and pleomorphic lipomas, while all angiolipomas and lipomas were nonamplified and eusomic. Thus fluorescence in situ hybridization is a sensitive, although not specific, tool for identification of spindle cell and pleomorphic lipomas.
Prognosis and Predictive Factors
Lipoma has an excellent prognosis.
12.7.2.2 Liposarcoma
Definition
Malignant mesenchymal neoplasm composed either entirely or in part of a mature adipocytic proliferation [79].
Epidemiology
Etiology
None.
Localization
Only 1 % of liposarcomas occur in the face. Our series contained only one liposarcoma.
Clinical Features
The patients present with painless or painful proptosis. The histologic subtypes (well differentiated (WDL), myxoid, dedifferentiated (DDL), and pleomorphic) are entirely separate diseases with different morphology, genetics, and natural history [116].
Macroscopy
Liposarcoma usually consists of large multinodular yellow masses that may contain solid, often tan-gray dedifferentiated areas. Dedifferentiated areas often show necrosis.
Histopathology
In contrast to benign lipoma, significant variation in cell size is appreciable. Focal adipocytic nuclear atypia and hyperchromasia is present. Hyperchromatic stromal cells tend to be more numerous within fibrous septa. A varying number (from many to none) of monovacuolated or multivacuolated lipoblasts and multinucleated cells (floret cells) may be found (Fig. 12.49). Floretlike cells, however, may be observed in in situ and prolapsed orbital fat [117]. Dedifferentiation is represented by the transition from liposarcoma to non-lipogenic sarcoma which, in most cases, is high grade.
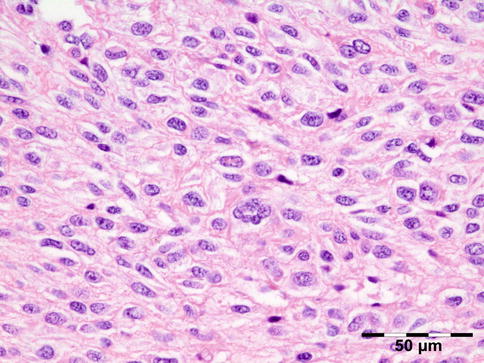
Fig. 12.49
Liposarcoma: this lesion is composed of plump to spindled cells; as is often the case no lipoblasts are present. Centrally a multinucleated floret cell is depicted (HE 400×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Many types of sarcoma.
Histogenesis
Mesenchymal stem cell with adipocytic potential.
Genetics
Both well-differentiated (WDL) and dedifferentiated liposarcomas (DDL) show amplification of MDM2. MDM2/chromosome 12 fluorescence in situ hybridization is a sensitive and specific tool in evaluating low-grade lipomatous neoplasms. However, p16 (Fig. 12.50) is the most sensitive and specific marker for detecting WDL/DDL, and the combination of immunohistochemical staining with CDK4 and p16 is of more discriminatory value than the combination of either with MDM2 (Fig. 12.51), the least sensitive and specific of the three markers [118].
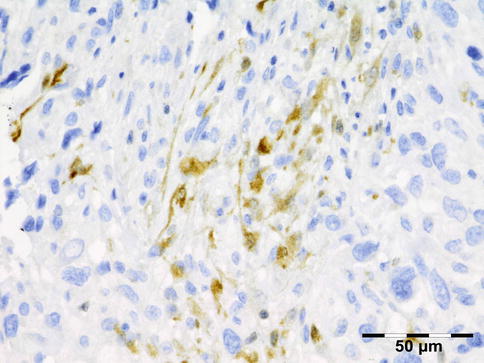
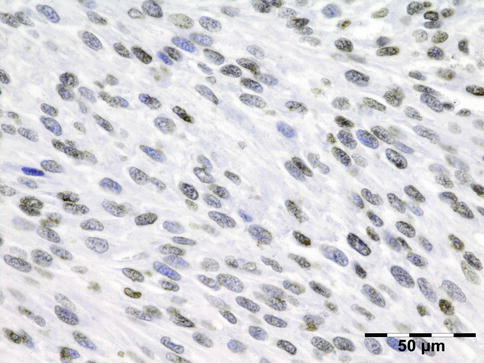
Fig. 12.50
Liposarcoma: positive cytoplasmic staining of the spindle cells for p16 is strongly suggestive for liposarcoma (p16 IH 400×) (Dr. Robert M. Verdijk)
Fig. 12.51
Liposarcoma: positive nuclear staining of the spindle cells for MDM2 supports the diagnosis of liposarcoma (MDM2 IH 400×) (Dr. Robert M. Verdijk)
Prognosis and Predictive Factors
Prognosis is related to the site and size and pathologic type and grade. Despite the difficulty in obtaining wide surgical margins, the small tumor size at presentation and the apparent predominance of the well-differentiated type mean that the prognosis for orbital liposarcoma is generally good [116].
12.7.3 Myogenic Tumors
12.7.3.1 Smooth Muscle Tumors
Leiomyoma
Epidemiology
Etiology
Posterior tumors are believed to originate from smooth muscle cells of vessel walls; anterior lesions may arise from the capsulopalpebral or Müller muscle.
Localization
Can be located anywhere in the orbit.
Clinical Features
They are well-encapsulated, slowly progressive solitary tumors that occur in the third and fourth decades.
Macroscopy
Leiomyomas are well-circumscribed, gray-white tumors (Fig. 12.52).
Fig. 12.52
Leiomyoma: gross examination shows a well-circumscribed, yellowish to gray-white tumor with a diameter of less than 2 cm (Dr. Robert M. Verdijk)
Histopathology
Tumors are composed of cells that closely resemble normal smooth muscle cells because they have eosinophilic cytoplasm with hematoxylin and eosin; fuchsinophilic, red-staining cytoplasm with Masson trichrome technique; and bland, uniform blunt-ended, cigar-shaped nuclei. They are arranged in orderly intersecting fascicles in a fibrous stroma rich in dilated sinusoidal capillaries (Fig. 12.53). They are highly differentiated and possess little or no atypia and, at most, an extremely low level of mitotic activity. Necrosis should not been present. Tumor cells are always positive for actin (Fig. 12.54) and desmin at least focally. S-100 protein is negative.
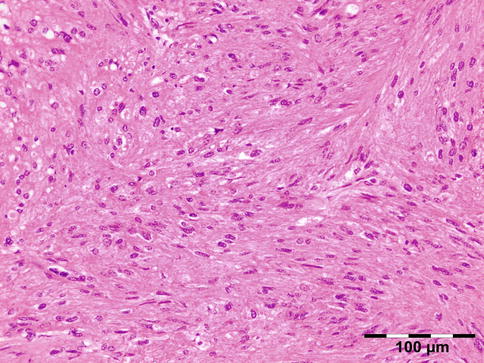
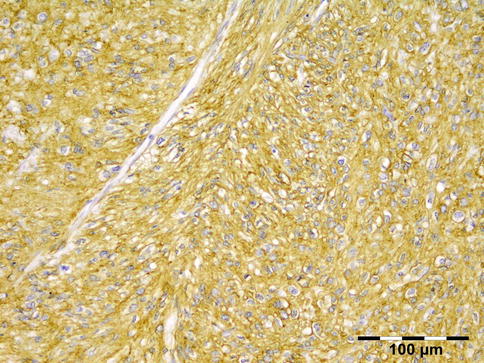
Fig. 12.53
Leiomyoma: the lesion is composed of a fascicular pattern of smooth muscle spindle cells with eosinophilic cytoplasm and bland, uniform blunt-ended, cigar-shaped nuclei (HE 200×) (Dr. Robert M. Verdijk)
Fig. 12.54
Leiomyoma: the lesional cells show a cytoplasmic staining for smooth muscle actin (smooth muscle antigen IH 200×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Nerve sheath tumors or fibrous histiocytoma.
Histogenesis
Smooth muscle cells.
Genetics
No genetic aberrations have been reported
12.7.3.2 Leiomyosarcoma
Epidemiology
Leiomyosarcoma is a rare orbital tumor; we encountered only one case in our series.
Etiology
Localization
May occur anywhere in the orbit.
Clinical Features
They are usually rapidly developing infiltrating masses.
Macroscopy
A fleshy mass, with colors varying from gray to white to tan. A whorled character may be evident. Larger examples may display hemorrhage, necrosis, or cystic change.
Histopathology
Histologic diagnosis rests upon demonstration of smooth muscle differentiation. The typical histologic pattern of leiomyosarcoma is that of intersecting, sharply marginated groups of spindle cells. This pattern may be less well defined in areas of some tumors, and occasionally there is a focal storiform, palisaded, or hemangiopericytoma-like arrangement. Fibrosis or myxoid change may be present. Hyalinized, hypocellular zones and coagulative tumor necrosis are frequent in larger leiomyosarcomas. The tumor cell nuclei are characteristically elongated and blunt ended and may be indented or lobulated. Nuclear hyperchromatism and pleomorphism are generally notable, although they may be focal, mild, or occasionally absent. Mitotic figures can usually be found readily, although they may be few or patchy, and atypical mitoses are often seen (Fig. 12.55). The cytoplasm varies from typically eosinophilic to pale and, in the former instance, is often distinctly fibrillar. Cytoplasmic vacuolation is frequently apparent, particularly in cells cut transversely. Epithelioid cytomorphology, multinucleated osteoclast-like giant cells, very prominent chronic inflammatory cells, and granular cytoplasmic change are unusual findings. Occasional soft tissue leiomyosarcomas contain areas with a nonspecific, poorly differentiated, pleomorphic appearance in addition to typical areas. Rarely, an osteosarcoma-like or rhabdomyosarcomatous component is associated with leiomyosarcoma. The use of immunohistochemistry with positive staining for desmin (Fig. 12.56), actin, and smooth muscle actin is of aid. S-100 protein is negative. Ultrastructural studies may be of help but have become largely superfluous with the advent of immunohistochemistry.
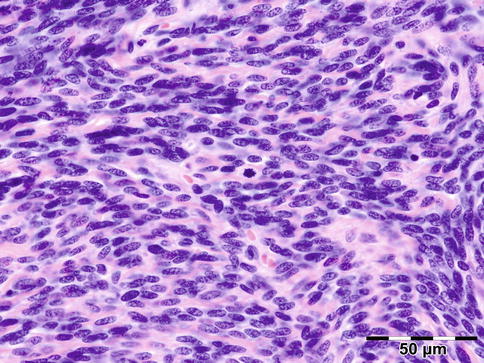
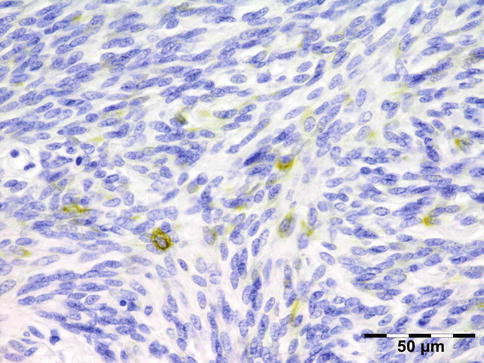
Fig. 12.55
Leiomyosarcoma: the lesion is composed of intersecting, sharply marginated groups of spindle cells. The tumor cell nuclei are elongated and blunt. Nuclear hyperchromatism and pleomorphism are mild. Mitotic figures can be readily observed (HE 400×) (Dr. Robert M. Verdijk)
Fig. 12.56
Leiomyosarcoma: sparse but distinct positivity for desmin can be observed in the tumor cells (desmin IH 400×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Sarcoma with leiomyomatous features.
Histogenesis
Smooth muscle cells.
Genetics
Most karyotypes are complex and no consistent aberrations have been noted.
Prognosis and Predictive Factors
May be treated with surgical excision combined with postoperative radiation.
12.7.4 Rhabdomyomatous Tumors
12.7.4.1 Rhabdomyoma
Definition
Extracardiac rhabdomyoma (RM) is a rare benign neoplasm of skeletal muscle differentiation that is classified into cardiac and extracardiac types. Extracardiac RM is further classified into adult (A-RM) and fetal (F-RM) types, depending on the degree of differentiation[79].
Epidemiology
In A-RM the median age is 60 years (range 33–80 years) with a 3:1 male predominance. In F-RM median age is 4 years (range, 3 days to 58 years) with a 2.4:1 male predominance.
Etiology
Unlike cardiac RM, there is no association with tuberous sclerosis.
Localization
RM occurs most often in the head and neck. Only five cases with occurrence in the orbit have been reported in the literature [125]. A-RM is often solitary (70 %) but may be multinodular (26 %) with discrete nodules in the same anatomic area. F-RM presents as a well-defined solitary mass involving soft tissue or mucosa.
Clinical Features
Progressive proptosis.
Macroscopy
The gross specimen consists of grayish-white tissue with hard fleshy consistency and some reddish components.
Histopathology
Reveals fetal or mature non-atypical rhabdoid differentiation.
Differential Diagnosis
Rhabdomyosarcoma.
Histogenesis
From rhabdomyoblastic cells.
Genetics
Some cases of F-RM are associated with nevoid basal cell syndrome.
Prognosis and Predictive Factors
Complete excision is the recommended treatment to prevent local recurrence.
12.7.4.2 Rhabdomyosarcoma (RMS)
Definition
Malignant soft tissue sarcoma that recapitulates the phenotypic and biological features of skeletal muscle [79].
Epidemiology
Etiology
The cause of most rhabdomyosarcomas is unknown.
Localization
The greatest proportion of tumors occur within the head and neck (about 47 %) and orbit (7 %) [128].
Clinical Features
About 42 % of patients with orbital rhabdomyosarcoma are aged 5–9 years; they present with proptosis or dysconjugate gaze [129]. Embryonal rhabdomyosarcomas constitute the most common subtype of rhabdomyosarcoma of the orbit by far. A special subtype of embryonal rhabdomyosarcoma is botryoid rhabdomyosarcoma (involving the conjunctiva) [130]. Alveolar rhabdomyosarcoma infrequently occurs in the orbit and may represent metastatic disease. Pleomorphic rhabdomyosarcoma is a high-grade sarcoma occurring almost exclusively in adults.
Macroscopy
Embryonal rhabdomyosarcomas form poorly circumscribed, fleshy, pale tan masses that directly impinge upon neighboring structures. Spindle cell rhabdomyosarcomas, like other spindle cell lesions, form firm, fibrous tumors with tan-yellow, whorled cut surfaces. Botryoid tumors, as the name implies, have a characteristic polypoid appearance with clusters of small, sessile, or pedunculated nodules that abut the epithelial surface of the conjunctiva. Alveolar rhabdomyosarcoma is so named because of the macroscopic impression of thin crisscrossing fibrous bands that appear as spaces between cellular regions of the tumor (reminiscent of lung alveoli).
Histopathology
RMS are composed of primitive mesenchymal cells in various stages of myogenesis, i.e., rhabdomyoblasts. Stellate cells with lightly amphophilic cytoplasm and central, oval nuclei represent the most primitive end of this spectrum. As these cells differentiate, they progressively acquire more cytoplasmic eosinophilia and elongate shapes, manifested in descriptive terms such as “tadpole,” “strap,” and “spider” cell. Bright eosinophilia, cytoplasmic cross striations, and multinucleation indicate terminal differentiation, and myotube forms may be evident (Fig. 12.57). Differentiation tends to be more evident following chemotherapy. The histologic architecture of embryonal rhabdomyosarcoma is typically composed of alternating areas of dense, compact cellularity and loose, myxoid tissues. The botryoid variant of embryonal rhabdomyosarcoma contains linear aggregates of tumor cells that tightly abut the conjunctival surface. This feature, known as a “cambium layer,” typifies these tumors which are extremely rare but may extend into the orbit. Densely arrayed whorls or fascicles of spindle cells constitute the spindle cell variant of embryonal rhabdomyosarcoma. These spindle cells often resemble smooth muscle cells, but cytoplasmic cross striations, if present, and/or bright eosinophilia indicates striated muscle differentiation. The presence of enlarged, atypical cells with hyperchromatic nuclei defines the anaplastic variant of rhabdomyosarcoma. This feature may be seen in both embryonal and alveolar tumors but is more prevalent in the former. Bizarre, multipolar mitoses are also often present. Anaplastic features can be focal or diffuse. Focal anaplasia indicates the presence of only single, dispersed anaplastic cells, whereas diffuse anaplasia indicates the presence of clone-like clusters of anaplastic cells.
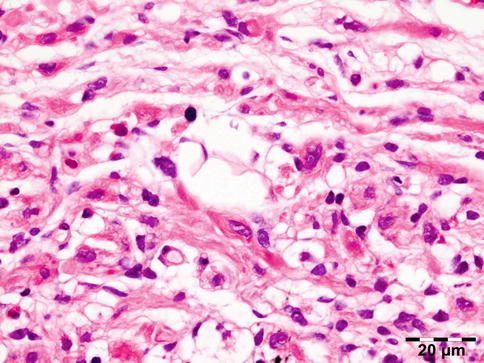
Fig. 12.57
Embryonal rhabdomyosarcoma: the lesion is composed of primitive mesenchymal cells with lightly amphophilic cytoplasm and central, oval nuclei, cells with more cytoplasmic eosinophilia and elongated shapes, “tadpole,” “strap,” or “spider” cell. Cells with bright eosinophilia, cytoplasmic cross striations, and multinucleation prove rhabdoid differentiation (HE 630×) (Dr. Robert M. Verdijk)
All alveolar rhabdomyosarcomas exhibit round cell cytological features reminiscent of lymphomas but with primitive myoblastic differentiation. Typical alveolar rhabdomyosarcomas produce fibrovascular septa that separate the tumor cells into discrete nests (Fig. 12.58). These nests contain central clusters of cells with loss of cohesion around the periphery. Tumor cells align the septa in a picket fence pattern. Giant cells with rhabdomyoblastic differentiation are common. Solid variant alveolar rhabdomyosarcomas lack the fibrovascular stroma and form sheets of round cells with variable rhabdomyoblastic differentiation (often little).
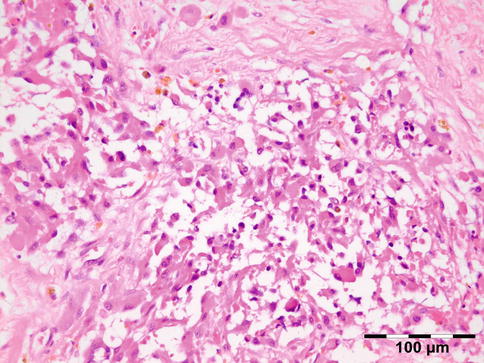
Fig. 12.58
Alveolar rhabdomyosarcoma: this metastatic lesion post chemotherapy shows fibrovascular septa that separate the tumor cells into discrete nests. The nests contain central clusters of cells with loss of cohesion around the periphery. Giant cells with rhabdomyoblastic differentiation are typically present (HE 200×) (Dr. Robert M. Verdijk)
Mixed embryonal/alveolar rhabdomyosarcomas contain foci with embryonal histology, i.e., myxoid stroma and spindle cell myoblasts as well as areas with alveolar histology. The alveolar foci usually contain nests with fibrous stroma, although highly cellular solid foci resembling lymphoma may occur.
Pleomorphic rhabdomyosarcomas are composed of bizarre polygonal, round, and spindle cells which display evidence of skeletal muscle differentiation [131]. No embryonal or alveolar component should be identified [132–134]. Cross striations are vanishingly rare.
Immunohistochemical evidence of at least one skeletal muscle-specific marker is required for the diagnosis of rhabdomyosarcoma. The presence of these markers correlates with the degree of tumor cell differentiation. Thus, only vimentin is present in the cytoplasm of the most primitive cells. Antibodies against MyoD1 and myogenin are highly specific and sensitive for rhabdomyosarcoma and are currently used as standard antibodies for diagnosis. Muscle markers such as desmin (Fig. 12.59) and muscle-specific actin (HHF-35) are shared by other cells with a myogenic phenotype. Occasional aberrantly expressed markers include cytokeratin, S-100 protein, neurofilaments, and B-cell proteins such as CD20 and immunoglobulin. The Pax5 protein can be present in alveolar RMS as a nuclear staining [135]. Smooth muscle actin and neuron-specific enolase staining occur rather frequently (in 10 and 30 % of rhabdomyosarcomas, respectively). With the use of immunohistochemical markers, the need for ultrastructural studies has become obsolete in many cases; however for the more challenging diagnoses, rhabdomyosarcomas exhibit a range of ultrastructural characteristics corresponding to those of developing striated muscle, primarily bundles of 5- and 15-nm thick and thin filaments punctuated by abortive Z-bands (Fig. 12.60).
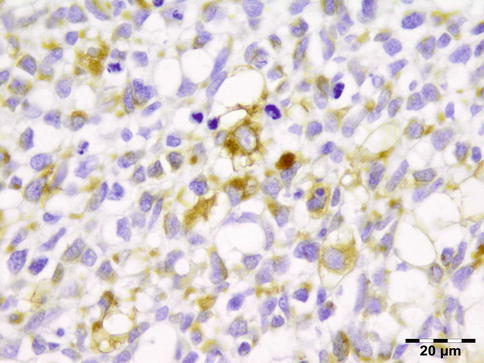
Fig. 12.59
Embryonal rhabdomyosarcoma: the rhabdomyoblasts, especially the more differentiated cells, stain positive for desmin (desmin IH 630×) (Dr. Robert M. Verdijk)
Fig. 12.60
Embryonal rhabdomyosarcoma: ultrastructural characteristics consist of bundles of five and 15-nm thick and thin filaments punctuated by abortive Z-bands that can be observed as irregular dense black dotted areas (EM) (Dr. Robert M. Verdijk)
Differential Diagnosis
Rhabdomyoma, other small blue round cell tumors.
Histogenesis
Rhabdomyoblasts.
Genetics
Embryonal rhabdomyosarcomas may result from sporadic or inherited mutations. Genetic syndromes that may be associated with rhabdomyosarcoma are neurofibromatosis (4–5 % risk of any one of numerous malignancies), Li–Fraumeni syndrome, Rubinstein–Taybi syndrome, Gorlin basal cell nevus syndrome, Beckwith–Wiedemann syndrome, and Costello syndrome. Cytogenetic studies of embryonal rhabdomyosarcoma have found complex structural and numerical chromosomal changes. Cytogenetic analyses can be used to demonstrate translocations that are consistently and specifically associated with alveolar rhabdomyosarcoma [136, 137]. Fluorescence in situ hybridization (FISH) is an easy tool to determine if translocations t(1;13) or t(2;13), associated with the alveolar subtype, are present (Fig. 12.61). In many centers, the use of RT-PCR to screen for a panel of translocations associated with soft tissue sarcomas is becoming a routine adjunct to morphologic analysis to help ascertain the diagnosis.
Fig. 12.61
Alveolar rhabdomyosarcoma: fluorescence in situ hybridization (FISH) is an easy tool to determine the presence of fusion signals of the translocation t(1;13) (FISH 400×) (Dr. Robert M. Verdijk)
Prognosis and Predictive Factors
Treatment responses and prognoses vary widely depending on location and histology. In patients with localized orbital disease, overall 5-year survival rates have improved to more than 80 % with the combined use of surgery, radiation therapy, and chemotherapy [129, 138]. Alveolar RMS has a worse prognosis when compared to alveolar RMS.
12.7.5 Pericytic (Perivascular) Tumors
Perivascular neoplasms comprise traditionally glomus tumor and hemangiopericytoma (HPC).
12.7.5.1 Glomus Tumor
Definition
Glomus tumors are mesenchymal neoplasms composed of cells that closely resemble the modified smooth muscle cells of the normal glomus body [79].
Synonyms
Typical glomus tumors are subcategorized as “solid glomus tumor,” “glomangioma,” and “glomangiomyoma” depending on the relative prominence of glomus cells, vascular structures, and smooth muscle.
Epidemiology
Glomus tumors are very rarely reported in the head region. Our series contained two glomus tumors; we are aware of three other reported cases of glomus tumors confined to the orbit [139].
Etiology
None.
Localization
This tumor is commonly found in the hand, where it often occurs as a small painful lesion beneath the fingernails. Although it may be present in many other locations, it is rare in the orbit [139].
Clinical Features
The patient may present with acute proptosis and ocular pain.
Macroscopy
Highly vascularized tissue.
Histopathology
Glomus cells are small, uniform, rounded cells with a centrally placed, round nucleus and amphophilic to lightly eosinophilic cytoplasm. Each cell is surrounded by basal lamina, seen best on PAS or toluidine blue histochemical stains (Fig. 12.62). Nests of glomus cells surround capillary-sized vessels. Glomangiomas, comprising approximately 20 % of glomus tumors, are characterized by dilated veins surrounded by small clusters of glomus cells. Glomus tumors of all types typically express smooth muscle actin and have abundant pericellular type IV collagen production. H-caldesmon is also positive. Other markers, including desmin, CD34, cytokeratin, and S-100 protein, are usually negative.
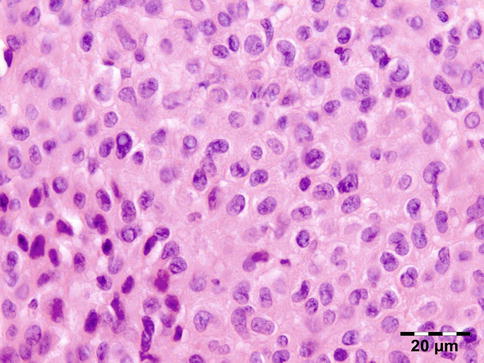
Fig. 12.62
Glomangioma: the lesion is composed of small, uniform, rounded cells with a centrally placed, round nucleus and lightly eosinophilic cytoplasm. Each cell is surrounded by a basal lamina (HE 630×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Hemangioma, smooth muscle tumors.
Histogenesis
Perivascular precursor cell.
Genetics
The genetic events underlying sporadic glomus tumors are not known. A gene for multiple inherited glomus tumors has been linked to chromosome 1p21-22.
Prognosis and Predictive Factors
The vast majority of the glomus tumors are benign, so they are treated by a simple surgical excision. A 10 % risk of local recurrence is related to an incomplete removal of the tumor. The malignant form of glomus tumor is exceptional.
12.7.5.2 Myopericytoma
The existence of HPC as a separate entity has been questioned because a number of neoplasms of different lines of differentiation are characterized by a HPC-like vascular growth pattern. Myopericytoma represents a recently delineated entity showing an HPC-like vascular pattern. Myopericytoma represents a distinct perivascular, myoid neoplasm of the skin and soft tissues, characterized by a broad morphologic spectrum of concentrically perivascular growing myoid tumor cells that stain positively for Alpha-SMA and often for h-caldesmon, whereas desmin is usually negative. Most cases of myopericytoma behave in a benign fashion, but local recurrences and rarely metastases may occur in atypical and malignant neoplasms [138]. All myopericytomas are located in subcutaneous and superficial soft tissue of distal extremities [140]. Myopericytoma in the orbit has so far not been described.
12.7.6 Vascular Tumors
12.7.6.1 Juvenile Hemangioma
Epidemiology
Our series contained seven capillary hemangiomas (11 % of all vascular lesions) confirming the fact that this tumor is common in the head and neck region [141].
Etiology
Used to be considered a hamartoma, that is, a congenital lesion composed of tissue normally present at the involved site. Now more likely considered to be neoplastic.
Localization
Often involves the periocular structures.
Clinical Features
The tumor usually presents in the first few weeks of life as an elevated cherry-red lesion involving the skin. The tumors initially are characterized by rapid diffuse growth for a period of 6–12 months, followed by a slow phase of natural resolution. Seventy percent of lesions resolve by the age of 7 years.
Macroscopy
The tumors are multinodular purple-red in color.
Histopathology
The histology is of capillary blood vessels that may vary from highly cellular areas with solid sheets of plump endothelial cells (Fig. 12.63) to clearly recognizable capillary vessels that may be accompanied by a progressive diffuse interstitial fibrosis (Fig. 12.64). Some mitotic activity usually is visible, but other features of malignant alteration are absent. The lesional cells stain positive for endothelial cell markers CD31 and CD34 and, in contrast to other hemangiomas, stain positive for GLUT-1, a marker normally present in retinal and CNS vasculature.
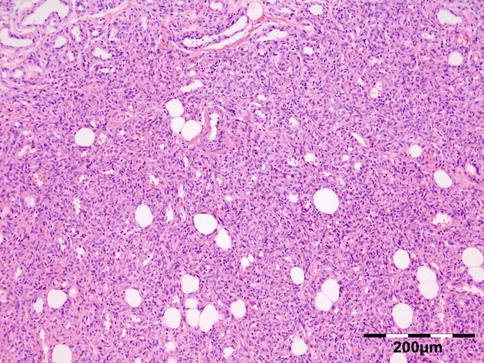
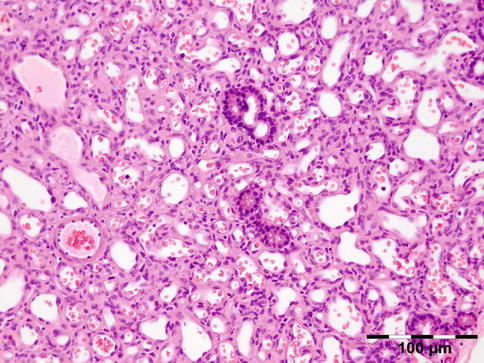
Fig. 12.63
Capillary hemangioma: this solid variant is composed of highly cellular areas with solid sheets of plump endothelial cells (HE 100×) (Dr. Robert M. Verdijk)
Fig. 12.64
Capillary hemangioma: this vascular type shows clearly recognizable capillary vessels and interstitial fibrosis. Some remaining ducts of the lacrimal gland can be seen (HE 200×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Clinical differential diagnosis is most challenging with rhabdomyosarcoma, lymphangioma, chloroma, and neuroblastoma. Histologic differential diagnosis is not very difficult.
Histogenesis
Vascular endothelial cell origin.
Genetics
Juvenile hemangiomas are monoclonal proliferations based on nonrandom X-inactivation. Some cases harbor somatic mutations of VEGFR2 or VEGFR3 indicating a neoplastic origin and not hamartomatous.
Prognosis and Predictive Factors
Operative treatment now is rarely performed with the advent of laser treatment and steroid injections, most recently combined by systemic beta-blockers. The tumor often has a diffuse growth and is difficult to remove.
12.7.6.2 Cavernous Hemangioma
Definition
Well-circumscribed low-flow vascular tumor composed of regular- and large-sized vascular spaces [79].
Epidemiology
Cavernomas (n = 37) are the most common type of vascular malformation of the orbit encountered in 56 % of vascular lesions and 3 % of all lesions in our series.
Etiology
Unknown.
Localization
Most lesions are located in the retrobulbar region, but they may occur anywhere in the orbit.
Clinical Features
Macroscopy
The tumor is usually well encapsulated and is often completely removed. The cut surface shows cavernous blood-filled channels with a spongelike appearance.
Histopathology
Shows large dilated blood-filled vessels lined by flattened non-atypical endothelium. The walls are often thickened by an adventitial fibrosis (Fig. 12.65). Mature bone is occasionally present.
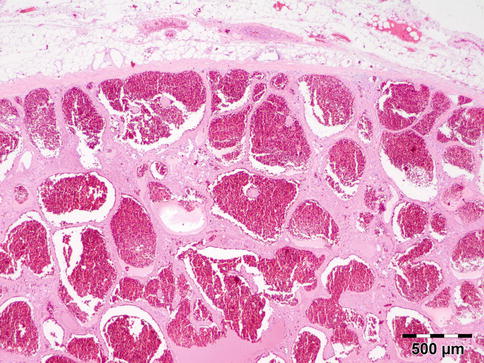
Fig. 12.65
Cavernous hemangioma: the lesion is well demarcated and composed of large dilated blood-filled vessels lined by flattened non-atypical endothelium. The vascular walls are thickened by adventitial fibrosis (HE 25×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Vascular malformations, lymphangioma.
Histogenesis
Hamartomatous vascular lesion.
Genetics
No genetic abnormalities have been reported with orbital cavernous hemangiomas.
Prognosis and Predictive Factors
Tumors causing symptoms are easily removed and no recurrences are observed.
12.7.6.3 Arteriovenous Hemangioma/Malformation (AVH/ AVM)
Definition
Nonneoplastic vascular lesion characterized by the presence of arteriovenous shunts [79].
Epidemiology
Deep-seated AVH is uncommon; our series contained 5 AVM out of 66 vascular lesions (8 %).
Etiology
Unknown.
Localization
Angiography demonstrates a high-flow angioma with feeders usually from the ophthalmic artery and external feeders from the maxillary artery.
Clinical Features
Deep-seated AVH affects children and young adults [74]. They are characterized by pulsating exophthalmos with evidence of bruit, occasional hemorrhage, thrombosis, or a caput medusae.
Macroscopy
Tumors are ill defined and contain variable numbers of small and large blood vessels, many of which are dilated.
Histopathology
AVH is characterized by large numbers of vessels of different size, which include veins and arteries with the former largely outnumbering the latter. Areas resembling a cavernous or capillary hemangioma are frequent, as are thrombosis and calcification. Combinations with lymphatic vessels can be observed (Fig. 12.66). Elastic stains are helpful in distinguishing between arteries and veins.
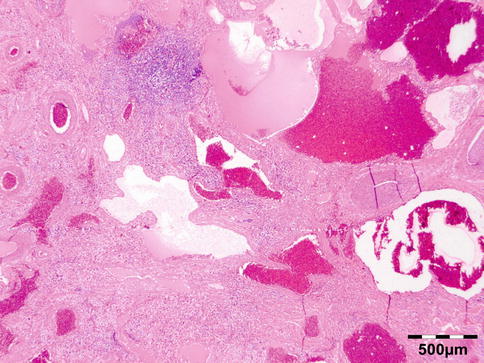
Fig. 12.66
Arteriovenous-lymphatic malformation: the lesion is characterized by vessels of different size, which include veins and arteries. Note that the veins outnumber the arteries. In this case a combination with lymphatic vessels can be observed (HE 25×) (Dr. Robert M. Verdijk)
Differential Diagnosis
This diagnosis always requires clinicopathological and radiological correlation.
Histogenesis
Hamartomatous vascular lesion.
Genetics
No genetic abnormalities have been described.
Prognosis and Predictive Factors
Treatment is difficult because of the degree of involvement, which has to be determined by angiographic examination. Local recurrence is common because of the difficulties in achieving complete excision [74].
12.7.6.4 Lymphangioma
Epidemiology
Lymphangiomas are common pediatric lesions. Lymphangiomas account for less than 10 % of vascular lesions and less than 1 % of all orbital lesions, depending on the case series. In our series, out of a total of 1,077 orbital lesions, 66 were vascular malformations (6 %), and of these, 5 (7.5 %) were lymphangiomas.
Etiology
Venous lymphatic malformations or lymphangiomas originate from the venous system and differentiate to lymphatic vessels.
Localization
Lymphangiomas may occur as intraconal as well as extraconal tumors.
Clinical Features
Lymphangiomas most often present at birth or during the first years of life. There is a tendency for the tumor to fluctuate in size with changes in posture, exertion and straining, and displacement of the eye. Secondary hemorrhage can lead to a sudden onset of painful proptosis [74].
Macroscopy
Multicystic or spongy mass, the cavities of which contain watery to milky fluid.
Histopathology
Lymphangiomas are characterized by thin-walled, dilated lymphatic vessels of different size, which are lined by a flattened endothelium and frequently surrounded by lymphocytic aggregates. The lumina may either be empty or contain proteinaceous fluid, lymphocytes, and sometimes erythrocytes (Fig. 12.67). Larger vessels can be invested by a smooth muscle layer, and long-standing lesions reveal interstitial fibrosis and stromal inflammation. Qualifying histopathological designations such as capillary, cavernous, or cystic are not currently used in the nomenclature of lymphangiomas since they are of no clinical relevance. Stromal mast cells are common and hemosiderin deposition is frequently seen. Positive staining for podoplanin (D2-40) is seen in lymphangiomas in contrast to hemangiomas. The endothelium demonstrates variable expression of FVIII-rAg and CD31 but usually not for CD34.
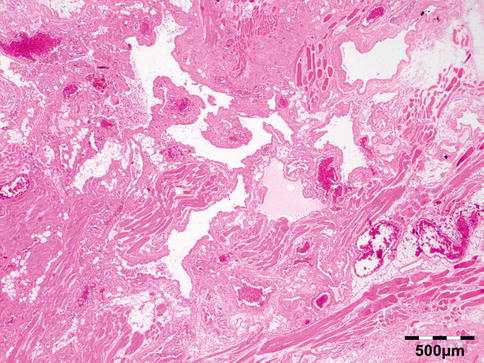
Fig. 12.67
Lymphangioma: the lesion is characterized by thin-walled, dilated lymphatic vessels of different sizes, which are lined by a flattened endothelium. The lumina contain proteinaceous fluid, lymphocytes, and sometimes erythrocytes (HE 25×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Hemangioma, vascular malformations.
Histogenesis
Hamartomatous vascular lesion containing lymphatic vessels.
Genetics
Cystic lymphangiomas (“cystic hygroma”) of the neck are often associated with Turner syndrome.
Prognosis and Predictive Factors
Treatment options for orbital lymphangiomas include conservative management; partial surgical resection of the major cyst; needle aspiration; intralesional injection of steroids, interferon, or sclerosing agents; and local radiotherapy. It is, in general, difficult to remove the orbital lymphangioma completely. They are not encapsulated, do not remain within anatomic boundaries, tend to penetrate into normal orbital structures, and can bleed profusely during surgery. New hemorrhagic cysts may form after incomplete resection. About half of the patients develop recurrences [74].
12.7.6.5 Epithelioid Hemangioma (Angiolymphoid Hyperplasia with Eosinophilia)
Epithelioid hemangioma is a benign vascular tumor with well-formed but often immature vessels, the majority of which are lined by plump, epithelioid (histiocytoid) endothelial cells. It is rare and represented only 1.5 % of all vascular lesions in our series. It presents as nodules or erythematous subcutaneous papules, usually in the head and neck region of young women [142]. It can occur in all races. Histologically, most lesions are well circumscribed and composed of vessels lined by plump endothelial cells that protrude into the lumen in a “tombstone fashion.” Surrounding the vessels, there is usually a prominent inflammatory infiltrate (Fig. 12.68). The endothelial cells stain positive for CD31 (Fig. 12.69). A large proportion of eosinophils can often be seen. Kimura’s disease is now known to represent separate entity [143] probably representing an allergic or autoimmune response that typically presents as subcutaneous nodules in the head and neck region of young Asian males [142]. There remains considerable controversy as to whether epithelioid hemangioma (angiolymphoid hyperplasia with eosinophilia) is a reactive lesion or a true neoplasm. Systemic associations include blood eosinophilia, nephrotic syndrome due to IgE deposition in the renal glomeruli, lymphadenopathy, and, less common, asthma, tuberculosis, and Loeffler syndrome. Complete local excision and follow-up are optimal management for epithelioid hemangioma. Local recurrence is reported to occur in up to one-third of patients.
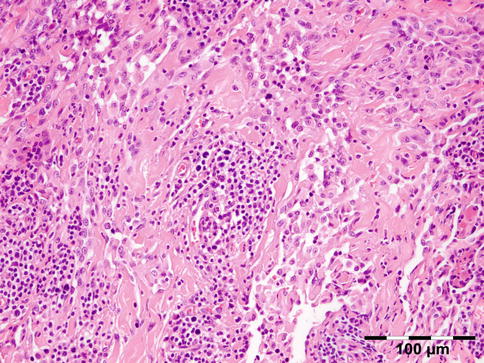
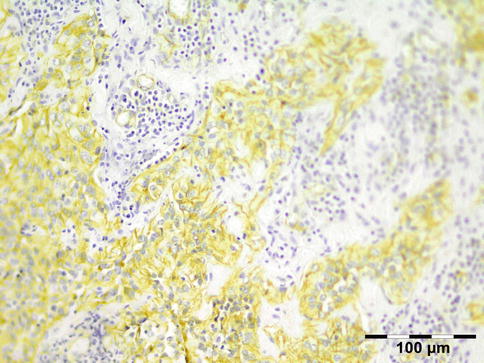
Fig. 12.68
Epithelioid hemangioma: the lesion is composed of vascular structures lined by plump endothelial cells that protrude into the lumen. Surrounding the vessels, there is a prominent inflammatory infiltrate with (in this case) occasional eosinophils (HE 200×) (Dr. Robert M. Verdijk)
Fig. 12.69
Epithelioid hemangioma: the epithelioid endothelial cells stain positive for CD31 (CD31 IH 200×) (Dr. Robert M. Verdijk)
12.7.6.6 Angiosarcoma/Epithelioid Hemangioendothelioma
Angiosarcoma accounts for less than 1 % of soft tissue sarcomas and has a predilection for the skin and superficial soft tissues [86]. Localization of angiosarcoma in the orbit is rare (only one case in our series), accounting for less than 3 % [144] of angiosarcomas (Fig. 12.70). Angiosarcoma may develop postirradiation. Epithelioid hemangioendothelioma is a vascular malignancy of endothelial cell origin, very rarely involving the orbit [145].
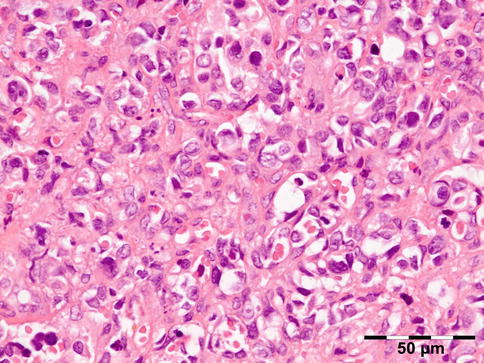
Fig. 12.70
Angiosarcoma: the lesion is composed of atypical epithelioid to spindle-like cells that form slit-like spaces filled with erythrocytes. Regularly cells show intracytoplasmic lumina with erythrocytes. The nuclei are polymorphic and polychromatic with mitoses (HE 400×) (Dr. Robert M. Verdijk)
12.7.7 Germ Cell Tumors
12.7.7.1 Teratoma
Definition
Teratomas contain tissue or tissues derived from all three primary germ layers: ectoderm, mesoderm, and endoderm.
Epidemiology
Rare orbital teratomas, embryonal carcinomas, endodermal sinus tumors (yolk sac carcinomas), and germinomas of the orbit have been reported [146]. We encountered two teratomas out of 1,077 lesions in our series.
Etiology
Teratomas belong to a class of tumors known as nonseminomatous germ cell tumor (NSGCT) [147]. Thus they are neoplasms as opposed to the historic designation as choristomas. In the orbit, teratomas arise from misdirected or ectopic germ cells [147], or they may secondarily involve the orbit originating from the intracranial or nasopharyngeal region [146].
Localization
No specific localization.
Clinical Features
Teratomas comprise approximately 1 % of orbital tumors in childhood. The congenital orbital tumor may be large at the time of birth. The preferred treatment is local excision.
Macroscopy
Pathologic examination discloses a circumscribed but not encapsulated, often multicystic, lesion.
Histopathology
Mature stromal elements may be composed of fat, cartilage bone, dermal elements, or glial tissue (Fig. 12.71). Mature epithelium can resemble epidermis and respiratory, gastrointestinal, or urothelial tissues (Fig. 12.72). Choroid plexus (Fig. 12.73) or retinal pigment epithelium may be present. Immaturity, usually manifest as immature neuroepithelium but sometimes additionally or rarely solely as cellular, mitotically active glia, is important to assess. Immature teratomas may be graded according to a system developed by Norris et al. [148] and modified by Gonzalez-Crussi [149]: grade 1 neoplasms contain immature neuroepithelium that involves no more than a single low-power (40×) field on the slide with the greatest amount of such tissue; grade 2 neoplasms contain immature tissue that involves more than one but not exceeding three low-power fields on any slide; and grade 3 neoplasms contain immature tissues that involve four or more low-power fields on any slide. Malignant transformation may occur after the development of the teratoma (“post-teratomatous” malignant transformation) [147]. This applies to those teratomas that develop malignant somatic neoplasms, such as sarcoma, a phenomenon reported thrice within the orbit [150–152], but also potential squamous cell carcinoma, adenocarcinoma, and malignant melanoma may develop. The prognostically most important malignant component to be identified in teratomas is yolk sac tumor which can lead to metastatic disease after complete surgical resection [153]. Immunohistochemistry shows staining patterns that are specific for the different tissue types that are reproduced. Minor yolk sac tumor components can be identified using a combination of α-fetoprotein and glypican-3.
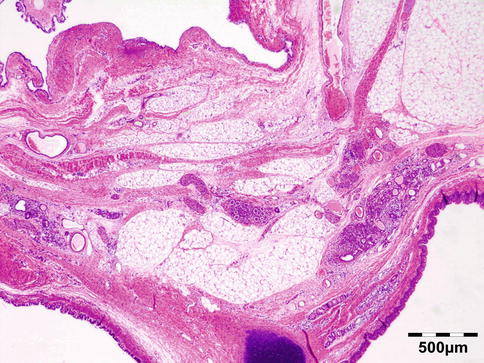
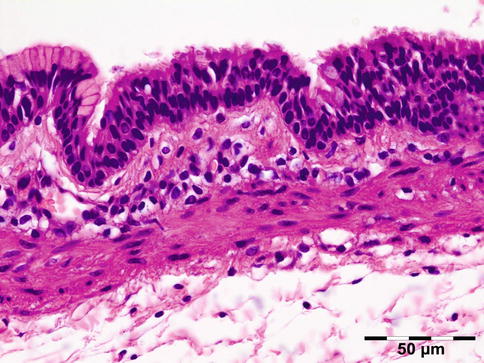
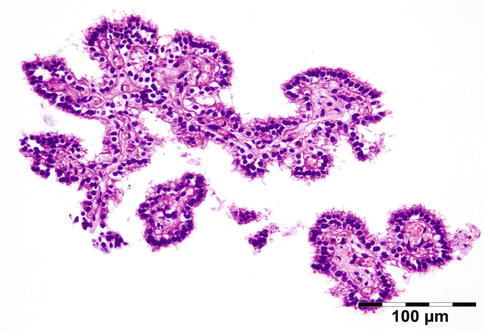
Fig. 12.71
Teratoma: mature stromal elements may be composed of fat, cartilage bone, dermal elements, or glial tissue (HE 25×) (Dr. Robert M. Verdijk)
Fig. 12.72
Teratoma: mature epithelium may resemble gastric/respiratory epithelium (HE 400×) (Dr. Robert M. Verdijk)
Fig. 12.73
Teratoma: choroid plexus (HE 200×) (Dr. Robert M. Verdijk)
Differential Diagnosis
Cystic lesions of the orbit.
Histogenesis
Non seminomatous pluripotent germ cell origin, gonocyte or primordial gonadal stem cell.
Genetics
These tumors are diploid with partially erased biparental imprinting. Gain of 12p is often present.
Prognosis and Predictive Factors
Teratomas of the orbit almost always are mature teratomas, but they may contain immature components [154]. Teratomas may be classified using the grading system adapted from Gonzalez-Crussi [149] by Norris [155]: 0 or mature (benign); 1 or immature, probably benign; 2 or immature, possibly malignant (cancerous); and 3 or frankly malignant. If frankly malignant, the tumor is a cancer for which additional cancer staging applies.
12.7.7.2 Yolk Sac Tumor/Endodermal Sinus Tumor
Definition
NSGCT with yolk sac differentiation.
Epidemiology
Rare.
Etiology
Like orbital teratoma, YST are NSGCT thought to arise from the yolk sac.
Localization
Clinical Features
They have a clinical presentation similar to embryonal rhabdomyosarcoma as aggressive tumors of early childhood.
Macroscopy
Hemorrhagic loose tissue fragments.
Histopathology
The tumor is composed of cords of cells that form a pseudopapillary pattern with a fine intervening stroma (Schiller–Duval bodies). The epithelial cells have anaplastic features and there may be myxomatous and necrotic areas. The tumor cells stain positive for alpha fetoprotein. Nuclear positive staining for OKT-3/4 is present.
Differential Diagnosis
Metastatic YST, metastatic carcinoma.
Histogenesis
Non seminomatous pluripotent germ cell origin, gonocyte or primordial gonadal stem cell.
Genetics
Not applicable.
Prognosis and Predictive Factors
Orbital lesions may have a relatively good prognosis due to early presentation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


