Fig. 10.1
Low-power histological section of the anterior part of the eye demonstrating the anatomical arrangements of the cornea, lens and anterior uvea (iris, ciliary process, ciliary body, pars plana and anterior choroid (×2 objective; HE) (Courtesy of Hardeep Mudhar, Sheffield, UK)
10.2 Embryology, Anatomy and Development
The iris is the anterior visible part of the uveal tract. The structure is named after the Greek goddess of the rainbow. The iris has two components: the posterior iris pigmented epithelium derived from neuroectoderm and the iris stroma derived from the neural crest (Fig. 10.2). Its major component, the iris stroma, is a loosely arranged tissue that contains pigmented and non-pigmented cells set in an abundant extracellular matrix containing bundles of type I collagen fibrils and hyaluronidase-sensitive glycosaminoglycans. The cells include melanocytes and fibroblasts. The greatest concentration of iris melanocytes is located in the avascular anterior border layer (Fig. 10.2). The iris vessels have a radial orientation and are ensheathed by a thick mantle of collagen fibres, giving a thick-walled appearance. The anterior surface of the iris has an irregular contour. This is because the papillary portion of the iris is thinner. Embryologically, this thinning results from atrophy of the anterior stroma caused by resorption of the papillary membrane. The anterior surface of the more peripheral iris is characterised by contraction furrows. Defects in the anterior stroma (crypts of Fuchs) are evident clinically. Heavily pigmented rounded clump cells of Koganei (either melanophages or neuroepithelial cells) are often found in the stroma near the sphincter muscle.
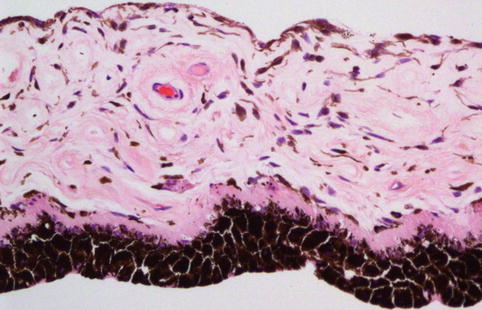
Fig. 10.2
Histological section of a normal iris, demonstrating the posteriorly located iris pigment epithelium, the adjacent dilator muscle, the loose iris stroma with thick-walled capillaries and the anterior accumulation of the normal melanocytes (×20 objective; HE)
The ciliary body is the middle part of the uveal tract, interposed between the iris and the choroid. It has two major components: the pars plicata (also known as the ‘corona ciliaris’) and the pars plana. The anterior pars plicata is composed of a ring of 70–80 ciliary processes, which project into the posterior chamber. Their inner non-pigmented epithelial cells secrete the watery aqueous humour that fills the anterior chamber. The aqueous leaves the eye by passing through the trabecular meshwork and its associated drainage channels located between the iris and the peripheral cornea. The ciliary processes of infants are smooth and pigmented. With age, the processes become more hyalinised as collagenous connective tissue accumulates around the vessels in their cores (Fig. 10.3a).
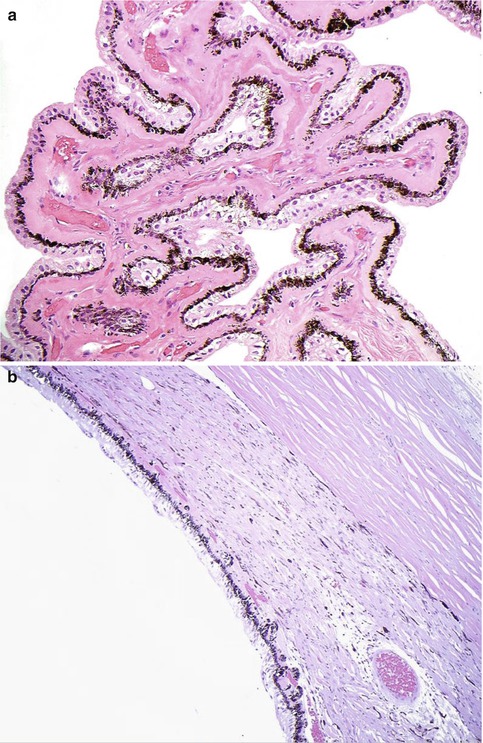
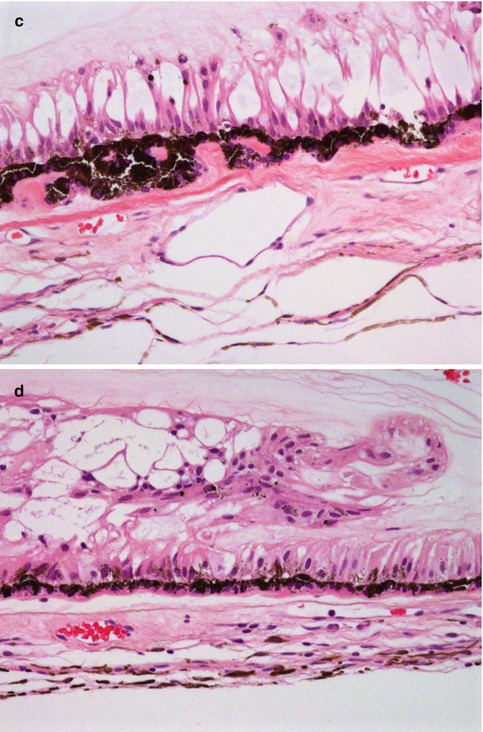
Fig. 10.3
(a) High-power magnification of ciliary body processes with the outer non-pigmented epithelium, the inner pigmented layer and the hyalinised stroma with the centrally placed blood vessels (×60 objective; HE) (Courtesy of Hardeep Mudhar, Sheffield, UK). (b) The pars plana is the flat part of the ciliary body and forms a circular band that extends to the ora serrata (×20 objective; HE). (c) Histological section of the ora serrata: the term ora serrata, meaning ‘toothed mouth’, is derived from the serrated appearance of this junction, caused by alternating dentate projections and oral bays of the peripheral retina (×60 objective; HE). (d) Peripheral microcystoid changes of the peripheral retina, giving it a ‘moth-eaten’ appearance (×60 objective; HE). (e) Histological section of the ciliary muscle, which has circular, longitudinal and radial parts, and its primary function is focusing or accommodation (×100 objective; HE) (Courtesy of Hardeep Mudhar, Sheffield, UK)
The pars plana (or flat part) of the ciliary body (Fig. 10.3b) is located posterior to the pars plicata and forms a circular band that extends to the ora serrata. The term ora serrata, meaning ‘toothed mouth’, is derived from the serrated appearance of this junction, caused by alternating dentate projections and oral bays of the peripheral retina (Fig. 10.3c). The dentate processes and oral bays are prominent in the nasal ora serrata. Posterior to the ora, the peripheral retina has a moth-eaten appearance due to the presence of peripheral microcystoid degeneration (Fig. 10.3d).
The outer layer of the ciliary epithelium is pigmented. At the ora serrata, it continues posteriorly as the retinal pigment epithelium (RPE), whilst the inner non-pigmented ciliary epithelium abruptly thickens to form the neurosensory retina. The stroma of the ciliary body is composed largely of smooth muscle. The ciliary muscle has circular, longitudinal and radial parts, and its primary function is focusing or accommodation (Fig. 10.3e). The longitudinal ciliary muscle of Bruecke attaches to the scleral spur, a ridge of connective tissue located directly behind the trabecular meshwork and the canal of Schlemm. The scleral spur is the only location where the ciliary body is firmly attached to the sclera.
The choroid is the posterior part of the uvea and is responsible for supplying oxygen to the outer layers of the retina. It is located between the RPE and the sclera, with Bruch’s membrane limiting it internally and the pigmented lamellae of the lamina fusca externally (Fig. 10.4). The anterior limit of the choroid is the ora serrata where the ciliary stroma merges irregularly into the choroidal stroma. Bruch’s membrane arises from a thin basal membrane of the pars plana pigment epithelium. The posterior part of the choroid wraps around the optic nerve and merges with its glial tissue and axons (Elschnig–Scheide or rim). Both Bruch’s membrane and the RPE also end close to the optic nerve edge. The thickness of the choroid varies with it being the thickest at the posterior pole, measuring 0.22 mm, and decreasing considerably to 0.10 mm at the ora serrata.
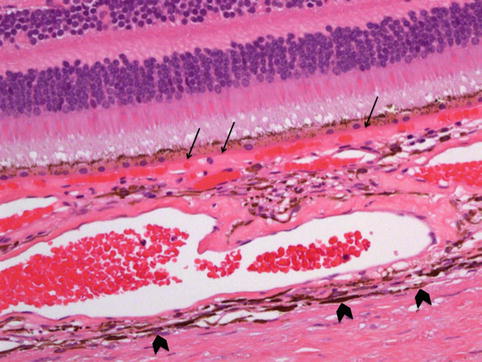
Fig. 10.4
Histological cross section of the choroid and adjacent retina, demonstrating its inner border, Bruch’s membrane (arrows) and the outer pigmented lamina fusca (arrow heads chevrons). It can be seen that the inner choriocapillaris contains the smaller capillaries of the lobules with the larger vessels being located externally. The sclera is the outermost coat of the eye (H&E section, ×40 objective)
The structure of the choroid is generally divided into four layers:
(a)
Haller’s layer – the outermost layer of the choroid consisting of larger diameter blood vessels
(b)
Sattler’s layer – the next inner layer comprising mainly medium diameter blood vessels
(c)
Choriocapillaris – the layer of capillaries
(d)
Bruch’s membrane (also termed lamina basalis, complexus basalis, lamina vitra) – the innermost layer of the choroid.
The choroid also consists of the choroidal stroma in which scattered lymphocytes and melanocytes are seen. It is thought that the melanin within the melanocytes aids the choroid limit uncontrolled reflection within the eye that would potentially result in the perception of confusing images.
The blood supply of the choriocapillaris arises from the posterior ciliary arteries, branches of which form the numerous end arterioles within the segments of the choriocapillaris. Each of the segments or lobules, which measure 620–830 nm, forms a miniature capillary system leading from a central blood vessel to the venules at the edge, which in turn drain into the vortex veins, numbering between 3 and 5 behind the equator of the eye. The endothelium of the choriocapillaris is fenestrated allowing for the permeability of substances, such as fluorescein. The choroid is loosely joined to the sclera, and hence there is a potential suprachoroidal space, which blends into the supraciliary space. Together both the supraciliary and suprachoroidal spaces extend from the scleral spur to Elschnig’s rim of the optic nerve. They are transected posteriorly only by the numerous posterior ciliary arteries and nerves.
10.3 Congenital Abnormalities
Congenital abnormalities of the uvea can be divided into those affecting the ‘uvea as a whole’, and those that affect one of the three structures within the uveal tract, particularly the iris.
10.3.1 Uvea (as a Whole)
(a)
Extramedullary haemopoiesis: In premature infants and less commonly in full-term infants, focal or diffuse extramedullary haemopoiesis may be observed. It is important to differentiate these from leukaemic infiltrates.
(b)
Congenital ocular melanosis: This is a unilateral condition caused by an increased number of atypical benign uveal melanocytes. The iris is darker and thicker than on the contralateral side, causing heterochromia iridum. The increased episcleral pigmentation renders the sclera a pale grey colour. This occurs in the pure ocular form of congenital melanosis and in the oculodermal melanocytosis of the naevus of Ota. White patients with congenital ocular melanosis also have an increased incidence of melanoma.
(c)
Albinism: The associated disturbance of melanin synthesis affects not only the uveal melanocytes but also the pigment epithelium [1, 2]. Histopathologically, there is an absence or a decrease of melanin pigment within the uveal melanocytes. The pigment deficiency is often complete within the melanocytes whereas pigmentation is decreased but still present within the melanin containing cells of the RPE. Many cases of albinism are associated with foveal aplasia, in which the foveal reflex is absent and no foveal depression can be demonstrated after serial sectioning.
(d)
Colobomas: These are caused by defects in closure of the embryonic ocular fissure, characteristically occurring inferonasally in the region of the fissure [3, 4]. Colobomas are defined as ‘conditions where a portion of the structure of the eye is lacking’. Because the primary defect in a typical coloboma is in the neuroectoderm, absence of the mesectodermal stroma is a secondary phenomenon. An absolute scotoma is present in the region of the coloboma because the overlying retina is absent or dysplastic. Although most colobomas are sporadic, they are occasionally inherited as isolated ocular defects, usually in an autosomal dominant fashion with incomplete penetrance.
10.3.2 Iris (Alone)
(a)
Aniridia: This is almost always a bilateral malformation that is inherited as an autosomal dominant trait (85 % of cases) or occurs sporadically [5, 6]. An association of the sporadic form of aniridia has been demonstrated to be associated with Wilm’s tumour of the kidney (Miller syndrome) and occurs in 13 % of cases. Most cases of aniridia are caused by mutations in the PAX6 gene on the short arm of chromosome 11. Miller syndrome is caused by deletions in 11p that include both the PAX6 gene and the tumour suppressor gene WT1 [7].
Both variations of aniridia can be associated with congenital or juvenile glaucoma. The designation ‘aniridia’ is actually a misnomer: these cases actually represent extreme hypoplasia of the iris, which are hidden clinically by opaque limbal tissues. Associated lens opacities and lens colobomas may occur, and foveal aplasia similar to that seen in albinism has been described.
(b)
Corectopia: There are several causes of corectopia (an eccentric location of the pupil), one of them being an anomalous closure of the embryonic fissure. Corectopia as well as polycoria (the presence of multiple papillary openings) can also be caused by primary dystrophic processes and by mechanical traction caused by the persistence of the anterior tunica vasculosa lentis and pupillary membrane.
(c)
Von Recklinghausen neurofibromatosis (NF-1): This is one of the most common hereditary diseases, as the NF-1 gene, which is located on chromosome 17, is quite large and is subject to mutation [10]. Neurofibromin is the protein product of the NF-1 gene and normally interacts with the ras oncogene to decrease growth stimulatory signals. The syndrome NF-1 is characterised by tumours of Schwann cells, which typically occur in the skin as multiple fibroma molluscum. Ocular findings in NF-1 are numerous, but include Lisch nodules (melanocytic hamartomas) on the iris and hamartomatous infiltration of the uvea with tactile corpuscle-like ovoid bodies [11].
(d)
Congenital iris stromal cysts: These are diagnosed in early childhood with most being recognised before the age of 10 years [12]. They have a thin wall and the lumen contains clear or slightly turbid fluid. Histopathologically, an iris stromal cyst is lined by thin non-keratinising stratified epithelium that sometimes contains goblet cells. It is thought that these develop secondary to developmental displacement of conjunctival epithelium into the iris during embryogenesis. Treatment options include aspiration, laser to the cyst wall, iridectomy or iridocyclectomy, mitomycin C as well as injection of alcohol into the cyst with irrigation.
(e)
Intraocular lacrimal gland choristoma: This is ectopic placement of lacrimal gland tissue within the eye, with most lesions affecting the eye, but some affecting both the iris and the ciliary body [13]. It appears in early infancy as a fleshy reddish pink mass with a slightly lobulated surface. Clear cysts can appear as a result of the ectopic lacrimal gland secretion and may progressively enlarge to cause iris atrophy, secondary glaucoma and cataract. Histopathological examination reveals normal lacrimal gland tissue. Whilst observation is recommended initially, local resection of the tumour is the treatment of choice.
10.4 Inflammation
Aetiology
Uveitis is often idiopathic, but it may be associated with systemic disorders including juvenile rheumatoid arthritis, ankylosing spondylitis, Reiter’s syndrome, ulcerative colitis, regional enteritis or Behcet’s disease. The reader is referred to specific reviews and textbooks, as the details are outwith the scope of the current chapter [14].
Histology
Broadly, uveitis can be divided into granulomatous and non–granulomatous disease. In the latter, microscopy reveals an infiltrate of plasma cells and lymphocytes in the uveal stroma. Sarcoidosis is the most common cause of granulomatous uveitis; close to 40 % of all patients with systemic sarcoidosis will have ocular disease at some stage of their disease [15]. The main differential diagnosis to granulomatous uveitis includes other autoimmune diseases (e.g. Wegener’s granulomatosis; Fig. 10.5a, b) as well as diseases of infectious origin (e.g. intraocular tuberculosis, leprosy, syphilis, parasites and fungal infections, including candidiasis and Lyme disease). It is important to note, however, that granulomatous inflammation is also found in sympathetic uveitis (see below) and Vogt–Koyanagi–Harada disease.
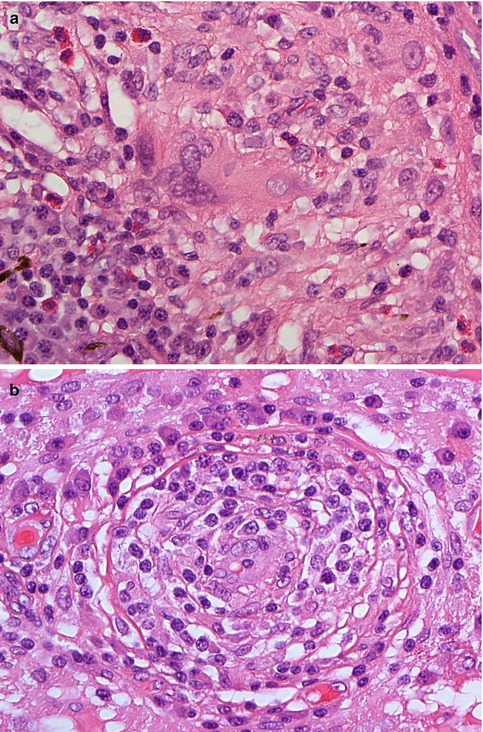
Fig. 10.5
(a) Histological section of a non-caseating granulomatous inflammation of the choroid with large multinucleate cells and scattered eosinophilic granulocytes in a patient with Wegener’s granulomatosis (×60 objective; HE). (b) Obliterative vasculitis of the choroid in Wegener’s granulomatosis (×60 objective; HE). Included in the differential diagnosis of such a case is Behcet’s disease, although granulomata are unusual in the latter
Special Inflammatory Subtypes
Vogt–Koyanagi–Harada disease (or uveomeningoencephalitic syndrome) is characterised by moderate to severe uveitis (anterior and posterior), secondary retinal separation, poliosis (whitening of hair), vitiligo (loss of cutaneous melanin), dysacousia and meningitis [16]. Not all patients have all symptoms and the degree of symptomatology differs from patient to patient.
Behcet’s disease is a systemic immune complex disease characterised by genital and oral aphthous ulcers and recurrent non-granulomatous iridocyclitis with hypopyon [17, 18]. Uveitis associated with obliterative vasculitis should raise the possibility of this disease.
Histiocytic inflammatory disorders can sometimes involve the uveal tract and include juvenile xanthogranuloma (JXG) and Langerhans’ cell histiocytosis.
JXG is an idiopathic benign inflammatory disorder of young children and occasionally adults [19]. It is characterised by multiple cutaneous yellow-pink papules that develop rapidly and resolve spontaneously without treatment. Most cases are confined to the skin, but involvement of ocular tissue, including the uvea, has been described [20]. On the other hand, many patients with intraocular JXG have no history of cutaneous lesions. Histopathologically, iris JXG presents as a variably sized mass with numerous small capillaries with an infiltration of histiocytes, lymphocytes, eosinophils as well as multinucleate giant cells of Touton type. The stain is negative for S-100P, excluding other histiocytoses such as LCH. JXG is usually responsive to steroids; other methods of treatment include iridectomy or iridocyclectomy.
LCH is a neoplastic proliferation of Langerhans’ cells with expression of CD1a, S100 protein and the presence of Birbeck granule on ultrastructural examination. It can be subdivided into (a) eosinophilic granuloma, (b) Hand-Schüller-Christian and (c) Letterer–Siwe disease. Ophthalmic involvement occurs in 10 % of cases, with the choroid being the most commonly involved ocular site (Fig. 10.6a–d). The differential diagnosis includes Rosai–Dorfman disease and necrobiotic xanthogranuloma [19, 21–23].
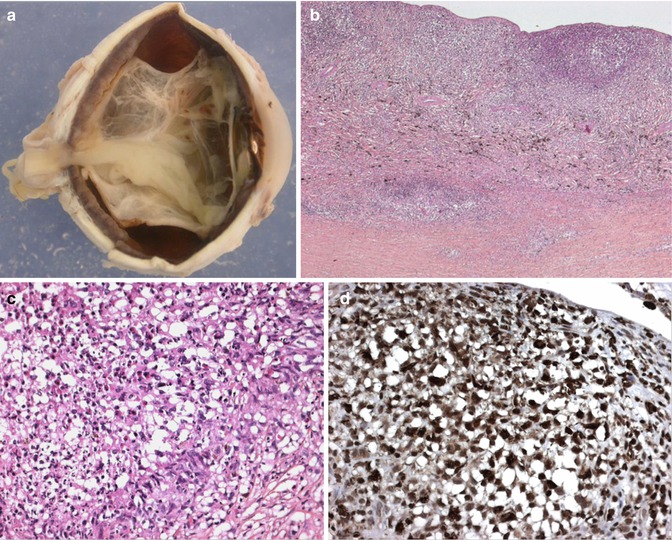
Fig. 10.6
(a) Macroscopic picture of an enucleated eye of a child with extensive uveal involvement by Langerhans cell histiocytosis. Thickening of almost the entire uveal tract is seen with an associated complete retinal detachment (×1.5 objective). (b) Low-power section of the choroidal thickening caused by a dense cellular infiltration confined to the choroid with some infiltration into the sclera (×10 objective; HE). (c) Higher power demonstrates that the infiltrating cells comprise a mixture of spindle cells with admixed eosinophils and occasional scattered multinucleate cells (×40 objective; HE). (d) The vast majority of the infiltrating cells demonstrated positivity for CD1a, S100 protein and CD68PG (picture) (×40 objective; PAP immunostaining) (Pictures courtesy of Dr Frederic Charlotte, Paris, France)
Treatment and Prognosis
This is dependent on the underlying cause of the inflammation and may require a battery of medications, ranging from glucocorticoid steroids to antimetabolite medications, such as methotrexate, depending on the degree of chronicity and aggressiveness of the inflammation [14]. The type of uveitis and its severity, duration and responsiveness to treatment or any associated illnesses all factor into the ultimate outcome of the disease.
The sequelae of ocular inflammation include corneal scarring and vascularisation, band keratopathy, cataract as well as secondary glaucoma. Intraocular fibrosis, gliosis and membrane (synechiae) formation are caused by attempts at ‘organisation’ of the inflammation. The end-stage findings can include, however, chronic retinal detachment, ocular shrinkage and hypotony, ultimately resulting in phthisis bulbi (atrophia bulbi with shrinkage and disorganisation). Osseous metaplasia of the RPE is often in the posterior parts of these phthisical eyes.
10.5 Injuries and Trauma
Introduction
The ophthalmic laboratory frequently processes eyes that have been subject to either surgical or nonsurgical trauma. Most cases show intraocular haemorrhage and loss or incarceration of intraocular structures. Injuries and trauma also predisposes to infection, which could be due to organisms introduced into the eye through penetrating injuries or contaminated intraocular foreign bodies, resulting in secondary exogenous endophthalmitis.
Severe contusions to the eye can cause separation of the iris, or iris and ciliary body, from the sclera (traumatic cyclodialysis). Trauma severe enough to do this also may cause retinal separation. This often leads to hypotony or phthisis bulbi. It also may be called recession of the chamber angle. It is not treatable.
The main condition to be discussed in this chapter is sympathetic uveitis (sympathetic ophthalmia).
Definition
Clinical Features
The patients present with blurred vision, photophobia and signs of inflammation in the uninjured fellow eye accompanied by signs of inflammation in the injured eye. Sympathetic uveitis occurs between 2 weeks and 1 year post-injury in about 90 % of cases, with most occurring in the 3-week to 3-month interval. It will not, however, develop if the injured eye is enucleated within 1 week of the injury. Delayed enucleation of the injured eye places the fellow at risk. Rare cases of sympathetic ophthalmia have been reported following ocular evisceration [26] and following proton beam in uveal melanoma [27].
Aetiology
Histology
Four characteristic features are found on histopathological examination of enucleated eyes with sympathetic uveitis. These include (1) diffuse thickening of the uvea by a granulomatous infiltrate composed of epithelioid histiocytes, inflammatory giant cells and CD8+ lymphocytes. Plasma cells are rarely seen in the infiltrate, and eosinophils can be found within the infiltrate in deeply pigmented patients. (2) The choriocapillaris is not destroyed by the inflammation, and the retina is spared. (3) The histiocytes usually contain granules of phagocytosed uveal pigment. (4) Nodular aggregates of epithelioid cells, which focally detached the RPE, are found on the inner surface of Bruch’s membrane [25]. These nodules, called Dalen–Fuchs nodules, are not pathognomonic for sympathetic uveitis, since they also occur in sarcoidosis and Vogt–Koyanagi–Harada disease.
10.6 Degenerative Conditions of the Uvea
Introduction
Degenerative conditions of the uvea are usually related to increasing age or as a complication of inflammation. Examples include:
(a)
Iris atrophy: This frequently occurs in previously normal irises as patients get older (senile iris atrophy). The effects on the pupillary border are most visible: the border will appear ragged and the sphincter muscle may be severely affected causing a loss of the pupillary response to light. This is important to remember when evaluating pupillary responses in an elderly patient. Histological examination demonstrates thinning of the sphincter muscle and of the iris stroma. Severe anterior uveitis or chronic glaucoma may result in destruction of the anterior uvea, particularly of the ciliary body. This eventually leads to phthisis bulbi or an enlarged, hypotonic globe, for which there is no treatment apart from enucleation.
(b)
Iris rubeosis: This is characterised by the formation of new fragile blood vessels on the surface of the iris, extending often into the chamber angle. This neovascularisation is usually associated with disease processes occurring in the retina, e.g. diabetes mellitus with advanced proliferative diabetic retinopathy, which results in the retina becoming ischaemic and releasing a number of angiogenic factors, most importantly vascular endothelial growth factor (VEGF). Other causes of retinal disease causing rubeosis iridis include central retinal vein occlusion, ocular ischaemic syndrome and chronic retinal detachment. It can also be seen following treatment of large choroidal melanomas that undergo the so-called toxic tumour syndrome (a term coined by Prof. Bertil Damato), following either proton beam or plaque therapy, releasing VEGF into the eye. This can be treated by removing the residual necrotic tumour tissue, e.g. by endoresection [28]. The blood vessels in iris rubeosis eventually undergo fibrosis, which may result in the formation of synechiae with the posterior surface of the cornea and in angle closure leading to ‘neovascular glaucoma’. Similarly, the iris leaf may be distorted and its papillary margin may be pulled posteriorly (rubeosis entropion uveae) or anteriorly (rubeosis ectropion uveae) (Fig. 10.7a).
(c)
Lacey vacuolation of iris pigment epithelium: This is due to the accumulation of glycogen and is present in 40 % of enucleated eyes from diabetics. It is termed ‘diabetic iridopathy’ (Fig. 10.7b).
(d)
Thickening of the ciliary body processes: This is particularly seen in patients with diabetes mellitus and can be highlighted in the periodic acid-Schiff (PAS) stain (Fig. 10.3a). The cause for this is unclear at present.
(e)
(f)
Choroidal neovascularisation membrane formation with breakdown of Bruch’s membrane is associated with a number of retinal diseases, including wet age-related macular degeneration (AMD). AMD is a very common cause of visual loss in older patients. It often produces haemorrhage that can sometimes simulate clinically a pigmented choroidal melanoma. The reader is referred to the numerous major reviews on AMD, as this disease is outwith the scope of this chapter [31–34].
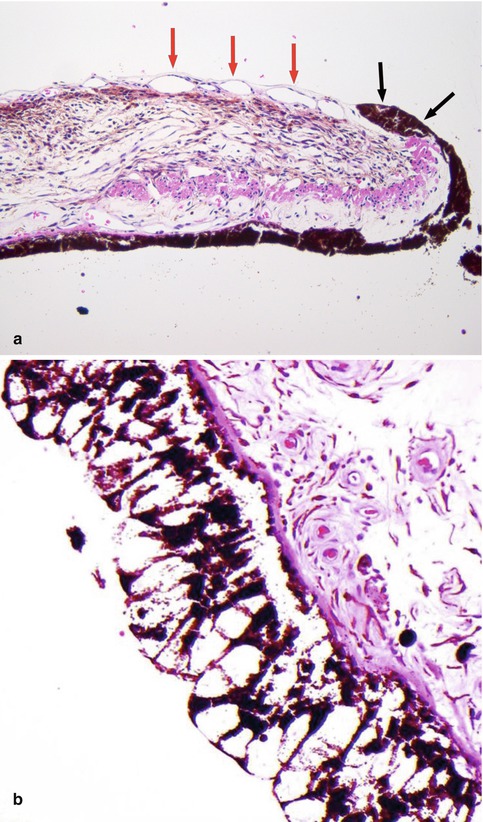
Fig. 10.7
(a) High-power histological section of an iris demonstrating ectropia (i.e. outward retraction of the iris pigment layer and adjacent sphincter muscle) (black arrows) caused by fibrosis of the fine layer of new blood vessels located on the anterior surface of the iris (red arrows). Such iris rubeosis leads to secondary neovascular glaucoma when extending into the anterior chamber angle (×60 objective; HE). (b) Lacey vacuolation of the iris pigment epithelium caused by the accumulation of glycogen within the cells, seen in 40 % of diabetic patients (Courtesy of Hardeep Mudhar, Sheffield, UK)
10.7 Neoplasms
10.7.1 Uveal Naevi
Definition
Uveal naevi are benign melanocytic neoplasms that do not demonstrate any cytological atypia and do not metastasise. They occur in 5 % of adults and are thereby the most common intraocular tumour.
Iris naevi present as pigmented macular lesions, are very slowly progressive and are often completely static over years. When the clinical presentation is not suspicious, in most cases the lesion will not be excised. Should the lesion be removed by, e.g., iridocyclectomy, histology shows a symmetrical well-defined lesion, located in the anterior part of the iris stroma, and usually composed of small spindle cells with small, uniform nuclei. Rounded naevus cells can be observed; epithelioid cells are exceptionally rarely seen and should be considered in the context of the surrounding tissue to differentiate the lesion from a melanoma.
Ciliary body naevi are very rare; the histology is comparable with iris naevi: spindle-shaped cells without atypia and without mitotic figures.
Most choroidal naevi typically appear as flat or minimally elevated patches of increased choroidal pigmentation that measure between 1 and 2 mm in diameter and <2 mm in height [28, 35]. Although they may be congenital, choroidal naevi are rarely observed in young children. They may be pigmented or amelanotic, and they can often have an irregular or feathery border. Choroidal naevi can cause the following secondary changes in the tissues surrounding them: (a) narrowing of the choriocapillaris, (b) RPE degeneration and proliferation with only tiny amounts of lipofuscin formation, (c) small serous retinal detachment with photoreceptor damage and (d) choroidal neovascularisation. Histologically, choroidal naevi are bland spindle cell tumours with the naevus cells having small oval- or cigar-shaped nuclei that lack nucleoli or nuclear folds (Fig. 10.8a, b). There is a variable degree of cytoplasmic pigmentation. Mitoses are absent. It is estimated that only 1/10,000–15,000 per year undergo malignant transformation to a melanoma [36, 37].
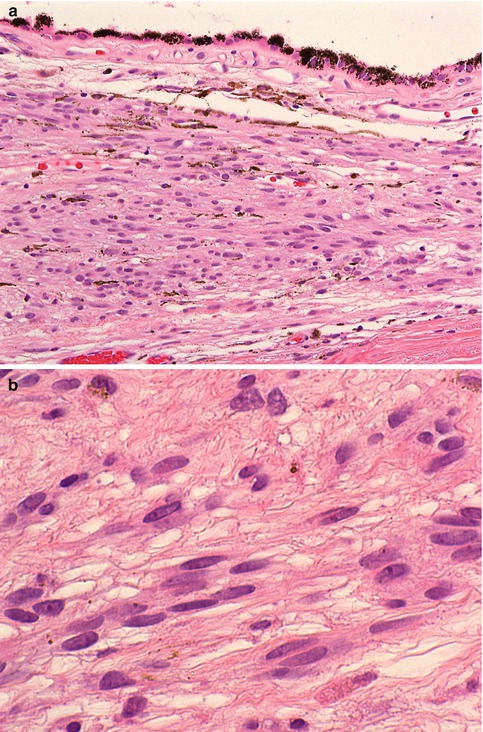
Fig. 10.8
(a) Histological section of a choroidal naevus (×40 objective; HE). (b) Higher power demonstrates that the naevus consists of loose stroma within which are spindle cells with elongated nuclei and very discrete nucleoli. Mitoses are absent (×60 objective; HE)
10.7.2 Melanocytoma
Introduction
Melanocytoma (syn. magnocellular naevus or hyperpigmented magnocellular naevus) is a characteristic type of uveal naevus, which merits a separate subsection [38, 39]. Melanocytomas classically involve the optic disc but can also occur in the iris, ciliary body and choroid. They are intensely pigmented and often occur in young patients, with a slightly higher incidence in females.
Clinical Features
Unlike uveal melanoma, it is rare in Caucasians and relatively common in dark-skinned people. For example, 37–50 % of optic disc melanocytomas have been described in African Americans. They may be quite large and difficult to distinguish from a melanoma on clinical findings. This is particularly the case if they extend into the anterior chamber and extraocularly, mimicking malignant invasion.
Histological Evaluation
Histological evaluation of melanocytomas usually requires bleached sections. These reveal a tumour composed of plump polyhedral naevus cells filled with large amounts of strongly pigmented cytoplasm and small discrete nuclei (Fig. 10.9a, b). Nucleoli are usually (but not always) inconspicuous. No mitoses are seen. Melanocytomas are known to undergo spontaneous necrosis, resulting in an infiltration of macrophages, which phagocytose the melanin. There may be associated pigment dispersion, which may reach the anterior chamber and cause obstruction to the drainage angles (melanocytomalytic glaucoma) [40].
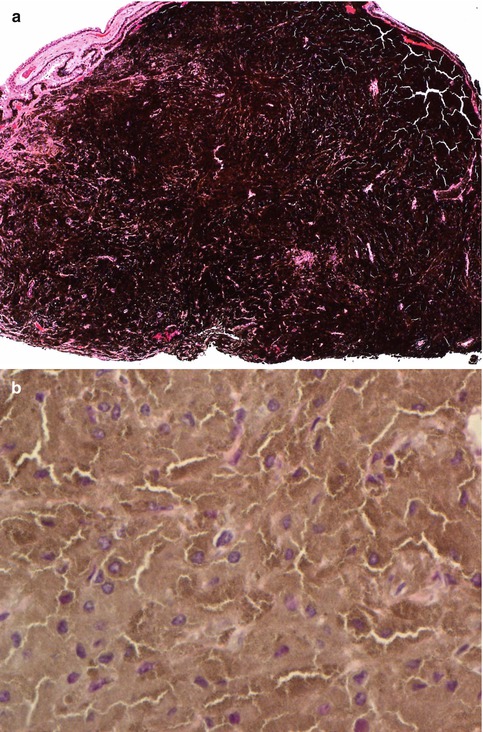
Fig. 10.9
(a) Iridocyclectomy specimen of a ciliary body melanocytoma, consisting of strongly pigmented naevus cells (×10 objective; HE). (b) The bleached HE sections demonstrate that the melanocytoma is composed of polyhedral naevus cells rich in cytoplasm and with small ovoid nuclei (×60 objective; bleached HE)
Genetics
Recent genetic examinations of two ocular melanocytoma have revealed GNAQ mutations in these two tumours [41].
Prognosis
Prognosis of melanocytoma for survival is excellent unless there is a very rare malignant transformation to melanoma [42]. This occurs rarely, with a frequency of <1 %. Visual loss may occur in optic disc melanocytomas, however, due to optic nerve damage (e.g. central retinal artery obstruction) and is usually permanent.
10.7.3 Bilateral Diffuse Uveal Proliferation (BDUMP)
Definition
Clinical Features
The most common malignant tumours associated with BDUMP are carcinomas, with ovarian and endometrial carcinomas being the most prevalent in women and lung and pancreatic cancer in men [46]. The patients present with gradual loss of vision and increasing intraocular pressure. On ophthalmoscopy, multiple red patches at the level of the retinal pigment epithelium, exudative retinal detachment and rapidly progressing cataracts are typically seen (Fig. 10.10a).
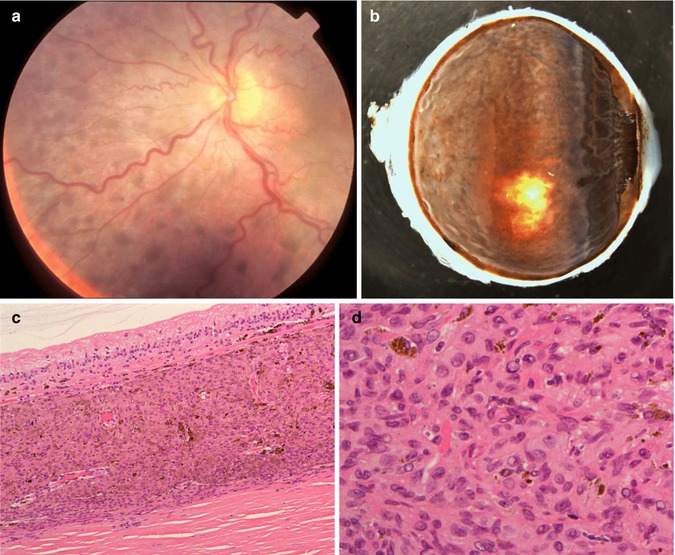
Fig. 10.10
(a) Fundoscopy of a 67-year-old woman with loss of vision in her right eye and with a history of a recently diagnosed clear-cell endometrial carcinoma. Multiple small nodular pigmentations within the choroid are seen (Courtesy of Prof. Damato, Liverpool Ocular Oncology Centre). (b) Enucleated right eye with a diffuse proliferation of pigmented cells within the uvea, also involving the iris (×1.5 objective). (c) Histological section demonstrating the diffuse cellular infiltration of partially pigmented cells limited to the choroid. Bruch’s membrane is intact. The overlying retina demonstrates atrophy (×40 objective; HE). (d) Higher-power magnification shows that the cellular infiltrate is composed of plump spindle cells with round oval nuclei and small nucleoli. No mitoses are seen. In the background are scattered melanophages (×60 objective; HE)
Aetiology and Genetics
The exact pathogenesis of BDUMP is unknown; however, many authors speculate that a humoral factor produced by the primary extraocular malignancy induces the melanocytes of the eye to proliferate. Recent genetic analyses demonstrate that the BDUMP cells do not show any changes in copy number for chromosomes 3 and 8 were found (by array comparative genomic hybridisation (CGH)), or GNAQ Q209 or DittoV600E mutations [47]. These findings are consistent with unpublished analyses of another case of BDUMP by Coupland et al. using multiplex ligation-dependent probe amplification (MLPA), examining copy number variation of chromosomes 1p, 3, 6 and 8 (unpublished; Fig. 10).
Histology
Histopathological examination of the specimens, usually enucleated eyes, shows diffuse infiltration of melanocytic cells in the choroid, ciliary body and iris root (Fig. 10.5b–d). These comprised a mixture of naevoid and spindle-shaped cells with very occasional cells displaying cellular atypia. No mitotic figures are observed. The cells stain positively with Melan A and HMB45.
Treatment and Prognosis
The management of BDUMP usually consists of treatment of the primary tumour, which should result in regression of the melanocytic proliferation within the uvea of the eye. Should, however, there be no initial response, low-dose external beam radiotherapy and anti-glaucomatous treatment ate considered. The prognosis of the ocular disease is dependent on the response of the primary tumour to therapy.
10.7.4 Uveal Melanoma
Definition
Uveal melanoma is by far the commonest primary intraocular malignancy in adults and is therefore discussed more fully than other tumours. It is a malignant tumour occurring predominantly in the choroid (90 %) with the remainder being confined to the ciliary body and the iris [28].
Epidemiology
The overall incidence of uveal melanoma is six to seven per million per year. It increases with age from about 2.5 per million between the ages of 15 and 44 years to 25 per million after the age of 65 years. The tumour is extremely rare in children. There is no significant sex difference. Uveal melanoma is much more common in Caucasians than in Africans or Asians. Epidemiological studies suggest that uveal melanoma is two to three times more common in blue/grey than in brown eyes. The importance of sunlight is uncertain but the evidence would suggest that it is unlikely to have a major role in uveal melanoma pathogenesis [28].
Aetiology
The aetiology of uveal melanoma is unknown; however, there are known associations with uveal melanoma. These include ocular melanocytosis (increased population of melanocytes within the uvea and episclera), oculodermal melanocytosis (naevus of Ota, which also involves the periocular skin and meninges), simple and dysplastic cutaneous naevi and cutaneous melanomas and neurofibromatosis type 1. Rare reports of families with an excess of uveal melanoma cases have been published. Recent evidence suggests that patients with a cancer susceptibility may have higher frequencies of uveal melanoma compared with the normal population [48, 49].
Localisation
Uveal melanomas arise in the choroid (80 %), ciliary body (12 %) and iris (8 %). Those occurring in the iris can be single nodular lesions, consist of multiple small nodules (tapioca iris melanoma) or extend as a ‘carpet-like’ growth across its anterior surface (Fig. 10.11) [28].
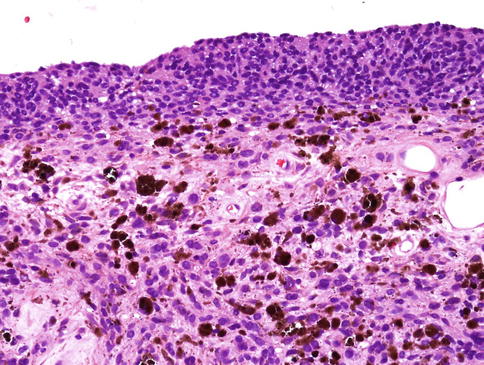
Fig. 10.11
Histological section of an iris melanoma of mixed cell type originating from the stroma but with diffuse spreading on the anterior surface (×40 objective; HE)
Clinical Features
Most frequently affected are white individuals in the sixth and seventh decades of life. Most choroidal melanomas are single and confined to one eye. The symptoms of patients with choroidal melanoma include:
(a)
Blurred vision caused by retinal detachment, posterior tumour location or vitreous haemorrhage
(b)
Visual field loss caused by retinal detachment
(c)
Floaters from vitreous haemorrhage
(d)
Photopsia described as a ball of light moving across the visual field over several seconds, more noticeably in subdued lighting
(e)
Pain secondary to neovascular glaucoma or uveitis
There is a wide range of therapeutic options for the treatment of primary uveal melanoma [28]. These include various forms of radiotherapy, surgical resection and phototherapy. The 5-year local tumour control rates in most specialised treatment centres exceed 90 %. Despite this, almost 50 % of patients with uveal melanoma will develop disseminated disease, predominantly in the liver, but also in the lungs (24 % of patients) and bone (16 %) [28].
Macroscopy
When performing the macroscopy of an enucleated eye with uveal melanoma, care should be taken to observe whether any extraocular melanoma is present. This can occur anteriorly (e.g. at the limbus) (Fig. 10.12a) or posteriorly via the emissary blood vessels or along the optic nerve. Transillumination in a darkened room usually will allow for localisation of the tumour. Sectioning should allow for a PO (pupil optic nerve) block that also includes the tumour. In early cases of choroidal melanoma, the sectioned tumour profile is oval- or almond-shaped. Although some melanomas diffusely infiltrate the uvea, most choroidal melanomas are well-circumscribed tumours with distinct margins. In many cases, the growing choroidal melanoma perforates Bruch’s membrane and pushes into the subretinal space where its apex typically assumes a collar stud or mushroom shape (Fig. 10.12b). Whilst a mushroom-shaped choroidal tumour is usually a choroidal melanoma, exceptionally rarely some metastases of systemic tumours can take on this configuration. The ruptured ends of Bruch’s membrane exert a compressive effect on the ‘waist’ of the choroidal melanoma causing vascular congestion and dilatation of blood vessels at the apex.
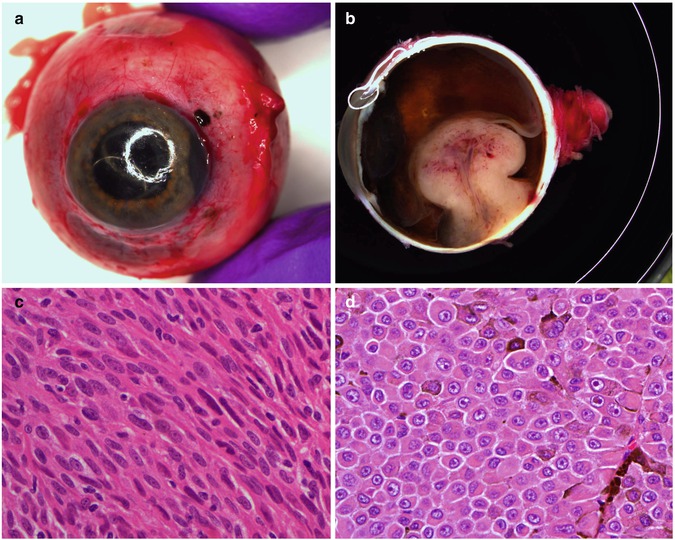
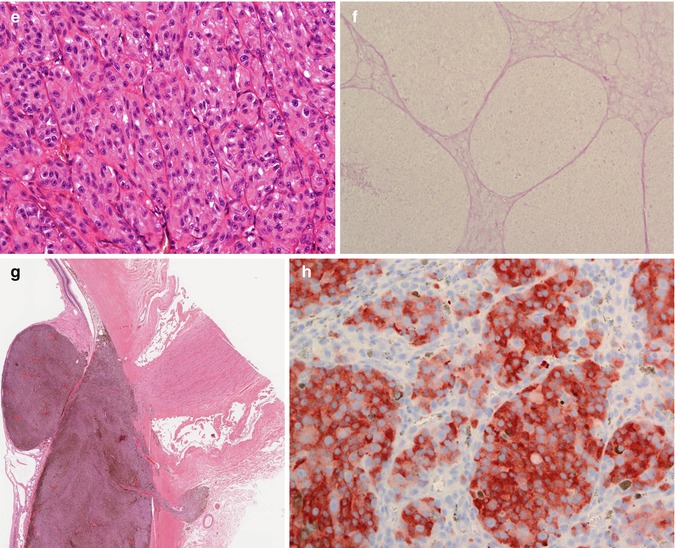
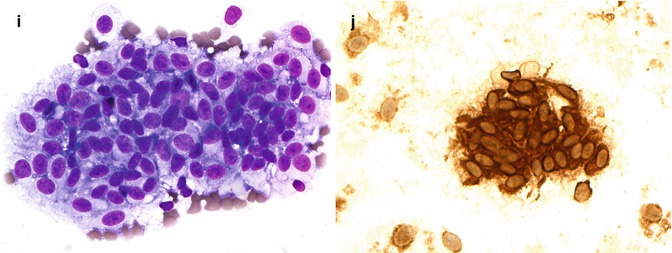
Fig. 10.12
(a) Gross examination of a fresh enucleated left eye shows a ciliary body melanoma on the lateral side of the eye with extension into the anterior chamber and with extraocular extension adjacent to the limbus at approx. the 1 o’clock position. (b) Sectioning of another fresh enucleated eye following transillumination demonstrates a non-pigmented choroidal melanoma with the classical ‘mushroom’ shape and central vascular congestion, as a result of Bruch’s membrane perforation. The overlying retina is detached. The lens is still in place. Reflection artefacts are present at the upper limbus and sclera. (c) Histological section of a spindle B-cell choroidal melanoma (×60 objective; HE). (d) Classical epithelioid cell choroidal melanoma with well-demarcated cell membranes almost with the suggestion of intercellular bridging across the artefactual cell gaps, and with voluminous eosinophilic cytoplasm with scattered melanosomes. Nuclei are large with prominent nucleoli. (e) Closed connective tissue loops in an epithelioid-celled ciliary body melanoma as demonstrated on HE (×40 objective). (f) PAS stain without haematoxylin counterstain highlights the closed connective tissue loops in another uveal melanoma (×20 objective). (g) Posterior extraocular extension in a comparatively small choroidal melanoma at the optic disc, with clear trans-scleral extension into the soft tissues adjacent to the optic nerve (×10 objective; HE stain). (h) Patchy HSP-27 staining in a choroidal melanoma of epithelioid cell type: loss of HSP-27 protein expression is associated with a loss of chromosome 3 [54]. (i) A cytospin of an intraocular biopsy of a choroidal melanoma of spindle cell type surrounded by a mantle of erythrocytes (×60 objective; MGG stain). (j) Positivity of the tumour cells in (i) for melanocytic marker, Melan A (×60 objective, PAP stain)
Choroidal melanomas typically cause an exudative serous retinal detachment of the overlying and adjacent retina. The overlying retina is often infiltrated by melanoma cells at the tumour apex, and dark vitreous seeding may be observed macroscopically.
Ciliary body melanomas are less common and tend to be more circular in shape, possibly with a ‘diffuse tail’ tapering posteriorly into the peripheral choroid. They may deform the crystalline lens and cause unilateral cataract. Occasionally, they may extend into the anterior chamber and seed along the iris surface or the trabecular meshwork, which may involve 360° of the anterior chamber (‘ring melanoma’).
Histopathology
Uveal melanomas consist predominantly of tumour cells with some admixed reactive cells usually representing a smaller proportion of the tumour’s cellular content. According to the modified Callender classification, uveal melanomas consist of spindle B-cells and/or of epithelioid cells (Fig. 10.12c, d) [50]. The former are long and fusiform with a small oval nucleus and a prominent nucleolus (Fig. 10.12c). Epithelioid, which means ‘epithelial-like’, cells are large and round with abundant eosinophilic cytoplasm, usually distinct cell margins and prominent nucleoli (Fig. 10.12d). Variants of epithelioid cells include wildly anaplastic tumour giant cells and relatively uniform small epithelioid cells. Spindle A cells are now regarded as benign and most commonly seen in uveal naevi (see above).
Notable histological features include:
(a)
Pigmentation of the melanoma cells – this can vary considerably between tumours and within tumours.
(b)
Lymphocytic and macrophage infiltration, which can be prominent and has been associated with a poorer prognosis.
(c)
(d)
Necrosis – varying degrees of necrosis may be found in uveal melanomas. It tends to be more prominent in rapidly growing choroidal melanomas or in tumours that have undergone brachytherapy. Aggregates of melanophages are typically in the necrotic areas. Total infarction of the tumour may results in destruction of adjacent tissues, including the sclera, possibly resulting in extraocular spread. The necrosis may lead to a ‘cystic’-like appearance to the tumour on ultrasound biomicroscopy.
Special stains of use include:
(a)
Masson-Fontana to label melanosomes in amelanotic melanomas
(b)
Permanganate or hydrogen peroxide bleach to allow cellular detail to be studied in deeply pigmented tumours
(c)
PAS stain without haematoxylin counterstain to analyse microvascular patterns or connective tissue loops (Fig. 10.12f)
Immunoprofile
Positive immunohistochemistry with S-100P, Melan A or HMB45 confirms the diagnosis in most cases. Proliferation markers such as Ki-67 and PC-10 provide an indication of cell turnover. Mitotic figures can be highlighted using PHH3 (also called Ser10) [53]. Recent work using proteomics and immunohistochemistry would suggest that decreased protein expression of HSP-27 is associated with genetic alterations of the uveal melanoma cells, correlating well with chromosome 3 status (Fig. 10.12h) [54].
Differential Diagnosis
The differential diagnosis of posterior uveal melanoma includes other benign and malignant neoplasms, including melanocytic naevi, choroidal haemangioma (see below) and metastasis from distant non-ocular primary neoplasms. The list includes other rare primary intraocular neoplasms that arise in the choroid such as schwannoma, leiomyoma, haemangiopericytoma and adenomas and adenocarcinomas of the RPE and the ciliary epithelium.
Non-neoplastic conditions that can simulate posterior uveal melanoma include vascular and haemorrhagic lesions (e.g. age-related maculopathy and peripheral exudative haemorrhagic chorioretinopathy), inflammatory and infectious conditions (e.g. nodular posterior scleritis, uveal effusion syndrome and granulomas) and a variety of miscellaneous disorders.
Histogenesis
Uveal melanoma is considered to be a cancer arising from uveal melanocytes. The precursors of melanocytes are non-pigmented melanoblasts derived from the neural crest, which bypass natural tissue barriers and basement membranes of the eye when migrating during embryogenesis. As with processes occurring in the skin, the melanoblasts mature into melanocytes within the uvea and/or give rise to melanocytic stem cells, which maintain the ocular melanocytic ‘system’. Although it is known that melanocytic stem cells reside in the hair bulge in the skin, the location of uveal melanocytic stem cells is still unknown [55]. It is hypothesised that various genetic and epigenetic alterations occur along the ‘melanoblast–melanocyte–naevus–uveal melanoma’ pathway, resulting in their malignant transformation and propensity to spread. It is unclear whether the ‘melanoma-initiating’ or ‘cancer stem-like’ cells, recently shown to be present in uveal melanoma cell lines, are derived directly from ocular melanocyte progenitors or from more mature melanocytes that have dedifferentiated.
Recent analysis of gene expression data of uveal melanoma would suggest that dedifferentiation indeed does occur during uveal melanoma development, but this requires further investigation. It is also unclear whether the naevus stage is a prerequisite in uveal melanoma development: it has been estimated that 1 in 8,000 naevi undergo malignant transformation to form uveal melanoma. Histologically, it is exceptionally rare for a residual naevus to be evident adjacent to or within a choroidal melanoma, supporting this observation.
Genetics
GNAQ and GNA11 mutations are also found in uveal naevi and in most uveal melanoma regardless of their tumour stage, chromosomal constellation or other outcome predictors (see below) [56, 57]. These mutations appear to be necessary but not sufficient for complete malignant transformation to melanoma. These data suggest that GNAQ and GNA11 mutations are early events in the molecular pathogenesis of uveal melanoma.
It has been known for almost 20 years that uveal melanomas show specific chromosomal alterations, which are quite distinct from melanomas at other sites, particularly those of the skin. The most striking abnormality in uveal melanoma is the complete or partial loss of chromosome 3. Other common genetic abnormalities of uveal melanoma include loss on 1p, 6q, 8 and 9p as well as gain on 1q, 6p and 8q [55]. These alterations were initially identified by standard karyotypic analyses. They have subsequently been confirmed by several groups using differing technologies, including fluorescence in situ hybridisation (FISH), CGH, spectral karyotyping, microsatellite analysis (MSA), MLPA and single nucleotide polymorphisms (SNPs) (see also review:[55]).
Prognosis and Predictive Factors
The above-mentioned chromosomal alterations in primary uveal melanoma are clinically relevant because of their correlation with the risk of metastatic death. Chromosome 3 loss is associated with a reduction of the 5-year survival probability from approximately 100 % to 50 %. Similarly, chromosome 8 gain and loss of chromosome 1 significantly correlate with reduced survival. Both chromosome 3 loss and polysomy 8q are also associated with other poor prognostic factors, including increasing tumour basal diameter, ciliary body involvement, presence of epithelioid cells, high mitotic count and closed connective tissue loops. Conversely, gains in chromosome 6p correlate with a good prognosis, suggesting that this aberration has a functionally protective effect [58, 59].
A PCR-based 12-gene assay based on gene expression profiling. The latter technique divides uveal melanoma into two ‘classes’ on the basis of an mRNA expression signature: class 1 and class 2. Class 1 uveal melanoma often show 6p and 8q gain. Class 2 uveal melanoma tends to show more aneuploidy with 1p loss, 3 loss, 8p loss and 8q gain [60, 61]. Class 2 uveal melanoma is also strongly associated with inactivating mutations of ‘BRCA1-associated protein-1’ (BAP1) located at 3p21 [62] (see also review [55]). Increasingly prognostic genetics testing is being performed on small intraocular biopsies (Fig. 10.12i, j). Essential for these analyses is a cytomorphological analysis of the cells being examined, as a ‘disomy 3’ or Class I result can potentially also be obtained when examining macrophages or indeed metastatic carcinoma.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


