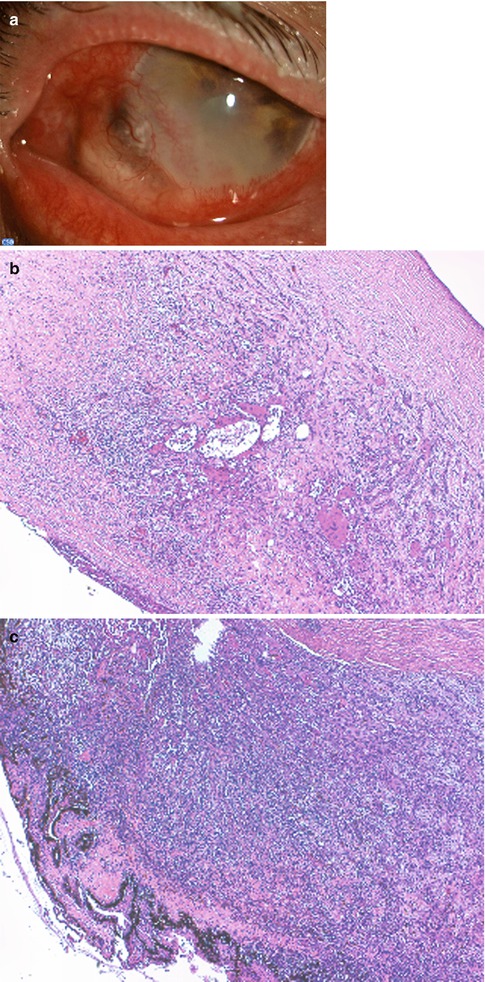
Fig. 2.1
Microphotograph of the eye (anterior segment) of a human fetus at the 42 mm stage (approximately 10th gestational week). (a) The conjunctival sac with its fornices is evident. Note that the eyelids remain fused, and they do not separate to form the interpalpebral fissure until after the 20th gestational week. HE stain. (b) Detail showing the conjunctival fornix lined by multilayered cuboidal epithelium. A few cells show intracytoplasmic vacuolization suggesting early formation of goblet cells. HE stain
Although the conjunctiva is a continuous membrane which only gradually changes its appearance, it is for practical purposes divided in three distinct zones: the bulbar, forniceal, and palpebral (tarsal) conjunctiva. The bulbar conjunctiva extends from the limbal region at the corneoscleral junction where it merges with the cornea. The loose connective tissue stroma of the conjunctiva is separated from the episclera by Tenon’s capsule composed of dense collagenous tissue. The conjunctival stroma is lined by stratified squamous, nonkeratinized epithelium. Further away from the limbus, goblet cells start to appear as individual cells lodged in the epithelium. Closer to the fornix, stromal inflammatory cells and lymphoid follicles become much more abundant. At the upper and lower fornix, the conjunctiva displays small folds to increase ocular mobility. The epithelium is markedly thinner than in the bulbar conjunctiva. The accessory lacrimal glands of Krause and Wolfring typically drain into the fornix. The conjunctival stroma is much reduced in thickness as the conjunctiva reflects onto the surface of the upper and lower eyelid. The number of goblet cells is reduced as the conjunctiva extends away from the fornix. The conjunctival epithelium is here markedly thin, featuring flattened cells at the mucocutaneous junction at the eyelid margin. The epithelial thickness increases markedly as the conjunctiva extends into the bulbar surface. The bulbar stroma contains abundant small blood vessels and lymphatics. At the limbus, there is a specialized, delicate network of small interconnecting perilimbal blood vessels. The connective tissue stroma of the bulbar and forniceal conjunctiva is only very loosely connected to the eye and episclera as this allows for unhindered ocular movement. Conversely, the palpebral conjunctiva is firmly fixed to the tarsal plate of the eyelids.
Nasally, the conjunctival sac contains the semilunar fold, a remnant of the nictitating membrane seen in animals. Further nasally, the semilunar fold (the shape resembling a half-moon) connects to the caruncle which represents a transition zone to the skin. As such, the caruncle often contains sebaceous glands, typical of the skin, but not seen in the conjunctiva proper. Rarely, the caruncle may contain apocrine glands. Occasionally, supernumerary caruncles may be present [2].
2.3 Congenital Abnormalities
Cryptophthalmos (from the Greek kryptos meaning hidden and ophthalmos meaning eye) is a unilateral or more often bilateral congenital anomaly characterized by abnormal eyelids and eye covered by intact skin. Typically, the conjunctival sac is absent and the eye(s) is microphthalmic. Cryptophthalmos may be part of Fraser’s syndrome, an entity defined by genital malformation and occasional malformation of the nose, ears, larynx, and renal system [3]. Fraser’s syndrome is an autosomal recessive disease linked to the FRAS1 and FREM2 genes and mapped to chromosome 4q21 [4]. Recently, mutation of the GRIP1 gene has also been associated with Fraser’s syndrome [5]. The incidence has been estimated to 0.043 in 10,000 live births [6].
Coloboma of the lid typically involves the palpebral conjunctiva as part of the defect. This congenital malformation may occur in association with an epibulbar choristoma and is sometimes a feature of the Goldenhar and Treacher Collins syndromes [7].
Hereditary hemorrhagic telangiectasia (Rendu–Osler–Weber disease) is characterized by systemic telangiectasia that may appear in the (often palpebral) conjunctiva and give rise to recurrent hemorrhages [1]. The Sturge–Weber syndrome (so-called nevus flammeus or angiomatosis associated with the trigeminal nerve branches) may have an ocular component characterized by diffuse choroidal hemangioma (essentially a hamartomatous malformation of the choroid) and episcleral and conjunctival vascular malformation. Importantly, children with Sturge–Weber syndrome may develop glaucoma and a number of other ocular complications [8]. Lymphangiectasia and lymphangioma are congenital vascular lymphatic malformations that may be cystic and circumscribed or more extensive and part of an orbital lymphatic malformation. These lesions may appear in any part of the conjunctiva, but are most common on the bulbar conjunctiva away from the limbus [9]. Typically, the cyst-like spaces are filled with clear fluid, but may sometimes, in association with trauma or without any apparent cause, be filled with blood and appear as hemorrhagic lymphangiectasia.
Choristomas contain ordinary cells not normally present on the site often as a result from a developmental abnormality. Conjunctival choristomas include the limbal dermoid often associated with Goldenhar’s syndrome. The limbal dermoid consists of hair shafts and sebaceous gland-like structures, believed to be displaced as a result of anomalous lid development. It typically occurs as a tan or pale dome-shaped structure at the inferior or temporal limbus [1]. Lipodermoids (alternatively termed dermolipoma) tend to present in the temporal periphery of the conjunctiva. These lesions are choristomas containing abundant fat (in contrast to limbal dermoids) and may fuse imperceptibly with the orbital fat. If excised they may present a surgical challenge because of the lack of a distinct cleavage plane. The rare epicorneal lipodermoid occurs centrally on the cornea and, in contrast to the limbal dermoid, appears not be associated with Goldenhar’s syndrome [10]. Complex choristomas typically consist of several cellular components, e.g., cartilage, bone (osseous choristoma), or lacrimal gland tissue (ectopic lacrimal glands) [11].
2.4 Inflammation
Acute conjunctivitis is usually bilateral and features vascular dilation, stromal edema, and abundant polymorphonuclear leukocytes. Inflammation may sometimes be extensive and cause the lining conjunctival epithelium to be ulcerated. Rarely, the acute inflammation induces stromal necrosis. Typically, acute conjunctivitis is caused by any of a large number of viral agents. Epidemic keratoconjunctivitis is associated with adenovirus infection and is an aggressive, highly contagious subtype including some distinctive clinical characteristics. This variant may be associated with tarsal membrane formation [12]. Disease usually resolves without sequelae, but corneal scarring may develop.
Chronic conjunctivitis has nonspecific characteristics like a mild to moderately increased number of stromal lymphocytes and stromal edema. Long-standing disease may generate additional findings. These include stromal fibrosis, depletion of goblet cells, and epithelial infoldings that may generate inclusion cysts that sometimes include small calcifications [1]. Papillae of the tarsal conjunctiva may also be present. This is a nonspecific finding that may, however, also be present in vernal conjunctivitis and giant papillary conjunctivitis [1]. Follicles are composed of accumulation of lymphoid tissue sometimes with a germinal center. They appear as small nodules, approximately 1 mm in diameter, and typically present in the fornix. The causes of chronic conjunctivitis include a host of infectious agents (Fig. 2.2), local irritants, chemical compounds, topical medications, foreign bodies, and even neoplasms [1]. Some agents cause a distinct appearance like the bilateral epithelial hyperplasia with follicles and papilla and in particular extensive whitish scarring seen in the upper tarsal conjunctiva after long-standing Chlamydia trachomatis infection. Over eight million people are estimated to have trachomatous trichiasis as a result of scarring leading to entropion, and trachoma of the eye remains the leading infectious blinding disease worldwide [13, 14].
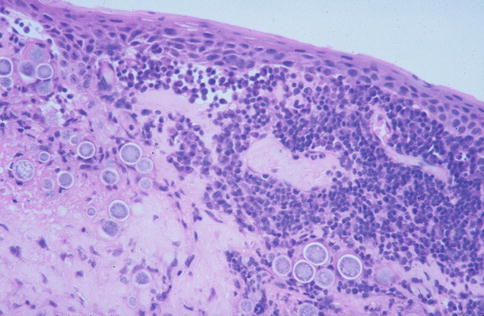
Fig. 2.2
Conjunctival coccidiomycosis. Numerous spherules about 60–80 μm in diameter and inflammatory cells are abundant in the conjunctival stroma. HE stain
Granulomatous conjunctivitis is uncommon and often unilateral. Pertinent histopathological features include chronic inflammation associated with caseating or non-caseating granulomas. Granulomatous conjunctivitis may arise in combination with systemic disease including Wegener’s granulomatosis and sarcoidosis [1, 15]. Occasionally, foreign bodies such as the hair shafts of caterpillars and infectious disease like onchocerciasis, blastomycosis, and tuberculosis may initiate granulomatous conjunctivitis [16–18]. Also, topical therapy including the use of preservatives has been associated with cicatrizing granulomatous conjunctivitis [19]. Secondary involvement of the tarsal conjunctiva may follow meibomian gland inflammation with or without the presence of a chalazion [20]. Rarely, cat-scratch disease may cause conjunctival involvement as a feature of the oculoglandular syndrome of Parinaud [1].
Allergic conjunctivitis is very common in the Western world (reportedly occurring in up to 15–20 % of the population) [21]. It typically presents as seasonal or perennial conjunctivitis in children and adolescents associated with itching. Vernal keratoconjunctivitis is a subtype, more common in the tropics than temperate climates, and presents in a limbal and palpebral form that may coexist. The limbal form is characterized by the so-called Tranta’s dots, but may also include giant papillae of the tarsal conjunctiva. Disease typically resolves by age 20 years. Atopic keratoconjunctivitis is another variant, often associated with atopic dermatitis. Histopathologically, the entity may feature giant papillae, but more commonly include conjunctival scarring. The conjunctiva is also prone to contact allergy caused by any of a range of low molecular weight substances. This reaction to a local irritant is cell mediated in contrast to the allergic variants listed above which are all IgE mediated [22].
Giant papillary conjunctivitis is caused by irritants and no longer believed to be an allergic reaction. These irritants include contact lenses, limbal sutures, ocular prostheses, and limbal dermoids [23, 24]. The histopathological features include the typical giant papillae of the upper tarsal conjunctiva (Fig. 2.3) and the presence of mast cells, basophils, and eosinophils. There is, however, no increase of IgE in the tear film as in true allergic conjunctivitis [22].
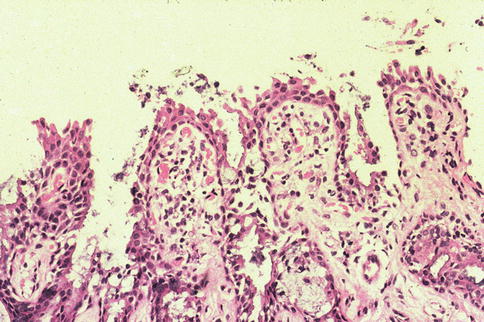
Fig. 2.3
Microphotograph of upper tarsal conjunctiva featuring giant papillae conjunctivitis with multiple infoldings. The stroma contains an increased number of chronic inflammatory cells and dilated blood vessels. HE stain
Hypersensitivity (phlyctenular) conjunctivitis is caused by proteins released by bacteria. In the Western world, this reaction to the eyelids and tarsal conjunctiva is often caused by hypersensitivity to staphylococcal infection. In a tropical setting, however, there is a significant association with coexisting tuberculosis [25]. Clinically, this entity is characterized by 2–3 mm whitish nodules composed of subepithelial acute and chronic inflammatory cells. These may cause an epithelial ulceration that eventually resolves by scarring [1].
Conjunctivitis may be associated with systemic disease, often dermatological disorders, like atopic keratoconjunctivitis (see above under allergic conjunctivitis), acne rosacea, mucous membrane pemphigoid, erythema multiforme (Stevens–Johnson syndrome), and epidermolysis bullosa [1]. Acne rosacea is a prevalent skin disease of middle-aged adults which frequently (58–72 %) is associated with manifestation in the eyelids and ocular surface [26]. The etiology and pathogenesis remain largely unknown. Skin involvement includes erythema, telangiectasia, papules, pustules, and phymatous changes in the cheeks, nose, chin, and central forehead. Changes may, however, be subtle, and acne rosacea may remain undiagnosed for prolonged periods of time. In some 20 % of patients, symptoms of ocular rosacea may precede skin symptoms [27]. Clinical signs suggestive of ocular rosacea include conjunctival and eyelid telangiectasia, eyelid and periocular edema, anterior blepharitis, and meibomian gland dysfunction [26]. Histopathologically, there is chronic conjunctivitis sometimes with the formation of papillae [27]. Disease intensity is variable, but keratitis and cicatrizing conjunctivitis may ensue [28]. Ocular pemphigoid (cicatricial pemphigoid) is a bilateral, albeit asymmetrical, autoimmune disease of the middle-aged and elderly. Diagnosis is made by immunofluorescent studies on conjunctival biopsies demonstrating basement membrane linear deposits of IgG, IgA, IgM, and complement 3 (C3) [29]. Sensitivity with this technique has been reported to range from 20 to 67 %, and a rebiopsy and/or additional techniques may be considered when a negative result is obtained in clinically suspicious disease. Standard histopathology is rarely conclusive often showing nonspecific features like subepithelial inflammation with lymphocytes, neutrophils, and histiocytes and reduced number of goblet cells. Occasional necrosis is accompanied by extensive stromal scarring. The characteristic subepithelial bullae are often not present or sometimes confused with the intraepithelial blisters occurring in pemphigoid. Ocular pemphigoid often runs an aggressive course and is a potentially blinding disease. A comprehensive review is available elsewhere [29].
Erythema multiforme (toxic epidermal necrolysis) and Stevens–Johnson syndrome are two of a group of acute blistering mucocutaneous disorders which often affects the conjunctiva [1]. Initiating events are usually drugs like allopurinol, anticonvulsants, and nonsteroidal anti-inflammatory drugs. Possibly, toxic epidermal necrolysis and the Stevens–Johnson syndrome are at two ends of a spectrum of disease. The average reported mortality for Stevens–Johnson syndrome is 1–5 % and 20–25 % for toxic epidermal necrolysis [30]. Disease may be preceded by fever, arthralgia, and malaise [1]. Clinically, the mucocutaneous lesions that follow are bilateral, multiple forms of vesicles and bullae combined with epidermal erosion (scalding). Histopathological examination of a skin biopsy reveals full-thickness epidermal necrolysis due to extensive keratinocyte apoptosis [30]. Scarring of the conjunctiva after healing may be extensive [1].
Epidermolysis bullosa is a rare, blistering mucocutaneous disease of autoimmune origin which may cause cicatrizing conjunctivitis. This entity is actually a heterogeneous group of disorders inherited in a dominant or recessive pattern. Epidermolysis bullosa disorders are caused by mutations of the structural proteins in the epidermis or dermoepidermal junction [31]. Currently, more than 1,500 different mutations in 17 different genes have been identified [32]. Diagnosis is made by a skin biopsy followed by immunofluorescence antigen mapping [31]. Transmission electron microscopy can detect the exact epidermal level of blistering and hence help to categorize disease into one of four groups now including more than 30 different subtypes. Signs of skin disease are often present shortly after birth and may be associated with involvement of the conjunctiva and other mucous membranes. Conjunctival vesicles, symblepharon, and chronic blepharoconjunctivitis as well as corneal involvement are hallmarks of ocular disease [1]. Intravenous immunoglobulin therapy may reverse the clinical course and improve ocular disease [33]. Disorders of the epidermolysis bullosa group are also candidates for protein replacement and gene therapy trials [34].
Ligneous conjunctivitis is a misnomer as the inflammation is secondary to extensive fibrin deposits, typically presenting in the upper tarsal conjunctiva. The surface epithelium is markedly thinned and atrophic or simply eroded. The surface is then covered by pseudomembranes largely consisting of cellular debris, fibrin, and polymorphonuclear leukocytes (Fig. 2.4). Ligneous conjunctivitis is caused by any of a range of mutations of the plasminogen gene. This causes functional plasminogen deficiency ultimately resulting in reduced fibrin clearance. Ligneous conjunctivitis may be associated with similar disease in, e.g., the mucous membranes of the airways and female genital tract [35].
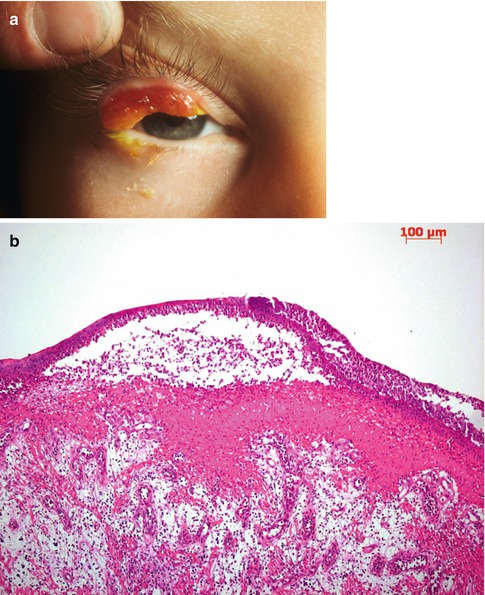
Fig. 2.4
Ligneous conjunctivitis. (a) The patient has characteristic pseudomembranes and tumorlike subtarsal thickening of the upper eyelid. (b) Fibrin is deposited in the conjunctival stroma. The deposit is lined by a partially detached pseudomembrane consisting of cellular debris and numerous polymorphonuclear leukocytes. Edematous granulation tissue appears beneath the fibrinous deposit. HE stain
2.5 Injuries
Thermal injuries may be caused by foreign bodies, exposure to cooled or heated air, flames, or fluids. Sometimes damage is iatrogenic, e.g., after the use of the triple freeze–thaw technique during removal of conjunctival tumors. The thermal response includes an initial phase of tissue destruction, an interval reactive phase, and a period of tissue repair [1]. Minor thermal injuries are typically limited to the conjunctival epithelium. Migration of peripheral epithelial cells then heals the ulceration with no scarring. Deeper injuries involve the stromal connective tissue and may cause a coagulation necrosis. The healing process will be prolonged and may result in significant scarring [1]. The extent of chemical injuries depends on exposure time and the ability to penetrate the conjunctival epithelium. Histopathologically, chemical injuries to the conjunctiva mimic the three phases listed above. Alkali burns are particularly feared, and late-phase excessive scarring may result in symblepharon and trichiasis secondary to entropion formation. Techniques to repair the ocular surface after both thermal and chemical injuries have much improved in recent years and include limbal stem cell grafting and amniotic membrane transplantation [36].
Aberrant wound healing may result in a vascularized, hypertrophic scar that mimics the appearance of a pterygium, but usually lack the stromal elastosis seen in pterygia. This scar may encroach on the corneal surface like a pterygium. Excessive wound healing may also cause a so-called pyogenic granuloma. This rapidly growing, protruding lesion is histologically defined by vascularized, edematous granulation tissue with abundant inflammatory cells (Fig. 2.5). The lining epithelium can be eroded, and pyogenic granuloma may bleed intermittently. Usually, an underlying lesion like a chalazion or a history of trauma, e.g., previous surgery, can be identified. An inclusion cyst is not an infrequent finding in the conjunctiva following trauma or surgery (Figs. 2.6 and 2.7). These cysts derive from epithelial cells being displaced into the stroma and then proliferating into stromal cysts. Inclusion cysts may reach a significant size. Usually translucent, they may contain cellular debris and chronic inflammatory cells. Retained foreign bodies may be embedded in the conjunctival stroma and then elicit a mild foreign body reaction with the presence of giant cells of the foreign body type (see below under granulomatous conjunctivitis). Rarely, pigment may dissolve and cause conjunctival pigmentation that may simulate a melanocytic neoplasia (see below under Sect. 2.7.5).
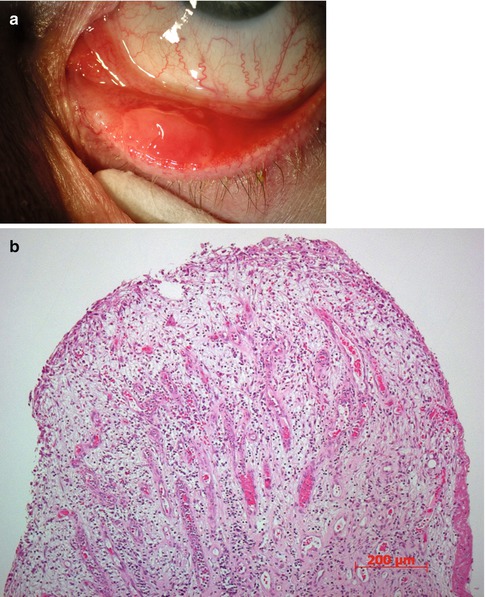
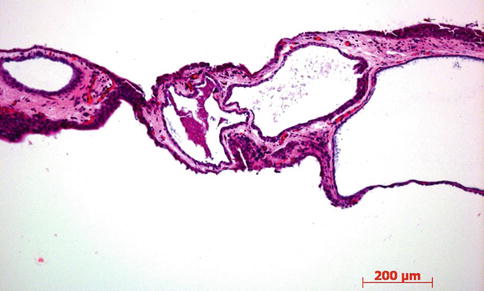
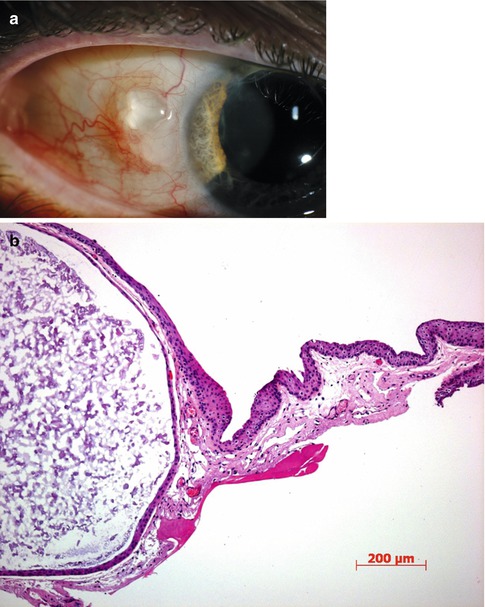
Fig. 2.5
Pyogenic granuloma. (a) Clinically, it arises from the tarsal conjunctiva close to the eyelid margin and lower eyelid punctum. (b) Histopathological examination shows dilated blood vessels, stromal edema, and numerous inflammatory cells. The epithelium is largely eroded
Fig. 2.6
Low-power microphotograph showing several conjunctival inclusion cysts after long-standing chronic conjunctivitis. HE stain
Fig. 2.7
Large conjunctival inclusion cyst. (a) Clinically, it is translucent and appears only to contain clear fluid. (b) Histopathological examination reveals the content to include desquamated cells and debris. HE stain
2.6 Degenerations
Conjunctival xerosis (drying of the conjunctiva) may be congenital (very rare) or acquired and then usually associated with vitamin A deficiency or Sjögren’s syndrome (keratoconjunctivitis sicca). Moreover, conjunctival xerosis may present following ectropion, proptosis, lacrimal gland dysfunction, and post-radiotherapy. Significant xerosis leads to metaplasia of the conjunctival epithelium with epidermalization including rete ridge formation and loss of goblet cells. The stroma displays abundant chronic inflammatory cells, blood vessel dilation, and vascular congestion [1]. Vitamin A deficiency remains a significant public health problem and globally causes a million instances of premature deaths and blindness every year [37]. Sjögren’s syndrome is still the preferred term used for the drying of the cornea and conjunctiva initially associated with polymyalgia rheumatica, but later with other connective tissue disorders like rheumatoid arthritis, scleroderma, and systemic lupus erythematosus [1]. The xerosis in Sjögren’s syndrome may affect mucosa at nonocular sites like the nasal, oral, and genital mucosa. Sjögren’s syndrome is presumably an autoimmune disorder.
The pingueculum is a circumscribed nodular yellowish lesion appearing nasally or temporally or both at the perilimbal conjunctiva in the interpalpebral fissure. The histopathological features include significant stromal collagen degradation (sometimes termed elastoid degeneration although there is no clear evidence that collagen has been replaced by elastin fibers), stromal hypervascularization, and edema. The pterygium appears as a flat scar-like tissue extending onto the corneal surface from the nasal conjunctiva and is usually clinically distinct from the pinguecula. The histopathological appearance is, however, quite similar to the pinguecula, apart from vascularization being more common and prominent in a pterygium. Both lesions are more prevalent in the elderly and in a tropical setting. For this reason and because of the histopathological appearance, both lesions are regarded as actinic, presumably caused by excessive exposure to ultraviolet radiation. When a pterygium extends onto the cornea, it may need to be surgically removed. Traditionally, this has been associated with frequent recurrence, but recent techniques using no sutures or intraoperative topical mitomycin have reduced this [38, 39].
Conjunctival amyloidosis is relatively uncommon and most often occurs as primary localized amyloidosis without systemic involvement [40]. Rarely, conjunctival amyloid deposits may be the first sign of systemic amyloidosis [41]. Typically, amyloid deposits are circumscribed, yellowish-pink, subepithelial mass-like lesions. Clinical features may also include blepharoptosis and recurrent subepithelial conjunctival hemorrhage. Biopsy will reveal subepithelial amyloid deposits that show apple green birefringence under polarized light and stain positive with the Congo stain (Fig. 2.8). Conjunctival amyloid deposits may show intrinsic vascularization [40]. Amyloidosis is caused by deposits of a group of abnormally folded proteins. In localized amyloidosis, deposits occur at the site of protein synthesis, and in systemic disease, deposits occur distant to the site of protein secretion. The reason for the protein misfolding that generates insolvable cross β-sheet structures is unknown, but may involve a combination and/or environmental factors [42]. Amyloidosis may be secondary to renal disease, inflammatory bowel disease, and malignancies, notably myeloma.
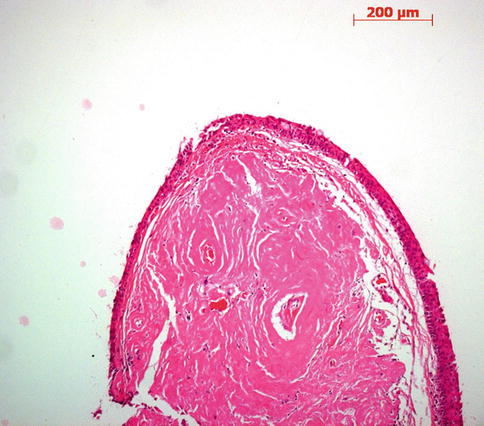
Fig. 2.8
Conjunctival amyloidosis. Abundant eosinophilic material is deposited in the conjunctival stroma. This stains positive with the Congo stain for amyloid and show birefringence in polarized light. HE stain
2.7 Neoplasia
2.7.1 Squamous Epithelial Lesions
2.7.1.1 Conjunctival Papillomas
Epidemiology
Papillomas are among the most frequent benign epithelial lesions arising within the conjunctiva of children or young adults.
Localization and Clinical Features
Preferred locations are the lid margin, interpalpebral conjunctiva, and caruncle. Most commonly they have an exophytic, sessile, or pedunculated appearance, with an irregular surface that sometimes may be cauliflower-like. When located at the limbus, they tend to remain flat.
Etiology
Various strains of human papillomavirus (HPV) have been identified in conjunctival papilloma. HPV types 6 and 11 are the most common types associated with conjunctival papilloma, and the HPV types 16 and 18 have also been found. p53 mutations in limbal epithelial cells, probably caused by ultraviolet irradiation, may be an early event in the development of some limbal tumors, including those associated with HPV.
Histopathology
Papillomas are composed of multiple fronds or fingerlike projections of conjunctival acanthotic epithelium that enclose cores of vascularized connective tissue (Fig. 2.9). Most lesions behave in a benign fashion and have little tendency to undergo malignant transformation; however, dysplastic changes can occur. Papillomas may have a mixed, or rarely an inverted, growth pattern.
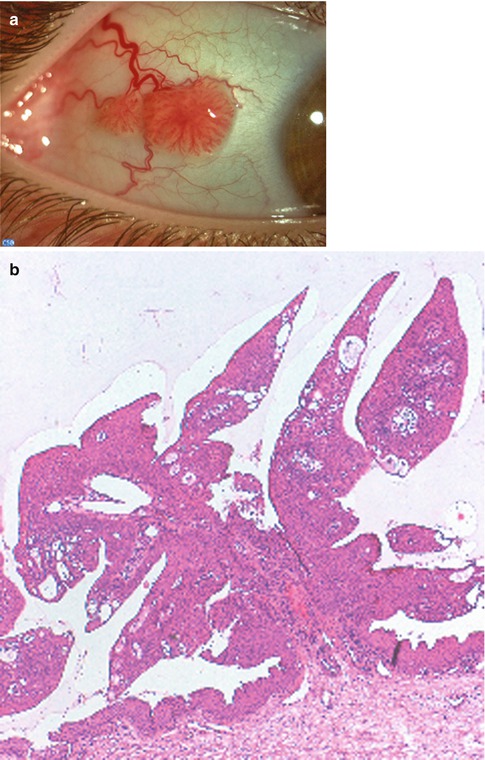
Fig. 2.9
Typical conjunctival papilloma. (a) Clinical appearance of an exophytic, sessile papilloma of the bulbar conjunctiva. (b) Histopathological features are those of fingerlike processes with a fibrovascular core covered by hyperplastic, nonkeratinized conjunctival epithelium. HE stain
The inverted conjunctival papilloma is a rare variant which typically has the propensity to invaginate into the underlying stroma instead of growing in the exophytic manner. Invasive lobules of benign epithelial cells are visible into the underlying connective tissue (Fig. 2.10). The presence of mucus-secreting cells is responsible for the term mucoepidermoid papilloma. Surgical excision with cryotherapy to the base and surrounding epithelium remains the most effective treatment [43–45].
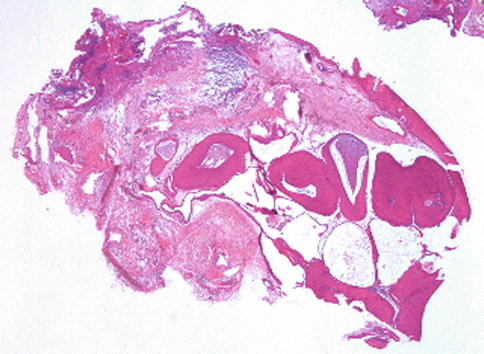
Fig. 2.10
Low-power histopathological features of an inverted papilloma. Lobules of epithelial cells have grown within the conjunctival stroma
Keratoacanthoma may be a specific variant of pseudoepitheliomatous hyperplasia, perhaps caused by a virus or more likely a low-grade type of squamous cell carcinoma.
Localization and Clinical Features
This rare, benign, solitary lesion presents as a white nodular mass, usually at the limbus, surrounded by dilated blood vessels and characterized by rapid enlargement.
Histopathology
The tumor forms central crater, with well-formed collarette, keratin filled, surrounded by invasive acanthotic epithelium exhibiting minimal cytologic atypia. The stroma may demonstrate elastotic degeneration and inflammatory infiltrate. Sometimes, the degree of atypia and invasive acanthosis may be more marked, causing difficulty in differential diagnosis from well-differentiated squamous cell carcinoma. Complete excision is recommended [1, 44, 46].
Hereditary benign intraepithelial dyskeratosis (HBID) is a rare autosomal dominant disorder affecting family members of the Native American Haliwa–Saponi tribe.
Localization and Clinical Features
The condition is characterized by bilateral elevated white to grayish epithelial plaques on the exposed perilimbal conjunctiva, associated with dilated episcleral vessels, sometimes with encroachment of the cornea. The conjunctival leukoplakic lesions are readily moveable over the underlying tissue. The epibulbar blood vessels are commonly hyperemic and have given rise to the common colloquial term for HBID: red eye disease. The disorder can also affect the buccal mucosa. HBID has an apparent seasonal variation, becoming worse in the spring and summer and subsiding in the cooler weather of autumn.
Histopathology
The histologic features of surgically excised lesions of HBID include hyperplastic stratified squamous epithelium with an overlying multilayered parakeratotic mantle containing rounded cells with dense eosinophilic cytoplasm and pyknotic nuclei. A lymphoplasmacytic infiltrate within the subepithelial stroma beneath the affected epithelium is typical.
Genetics
A duplication in chromosome 4q35 is associated with HBID.
Prognosis
Keratotic plaque. Leukoplakic lesions with no potential for malignant transformation may develop in the bulbar or limbal conjunctiva. Histologically, these lesions lack the dyskeratotic features of actinic keratosis, and the epithelial thickening is characterized mainly by acanthosis and parakeratosis or orthokeratosis [1].
Actinic keratoses are focal leukoplakic lesions, located near the limbus in the interpalpebral area. The lesions develop slowly and often occur within the epithelium overlying a preexisting pinguecula or pterygium. Microscopically there are irregular focal acanthosis of the epithelium by atypical epidermoid cells and a surface plaque of parakeratosis. The actinic elastosis in the substantia propria is considered as one of the characteristic features of this precancerous condition related to prolonged exposure of the conjunctiva to ultraviolet light (Fig. 2.11) [43].
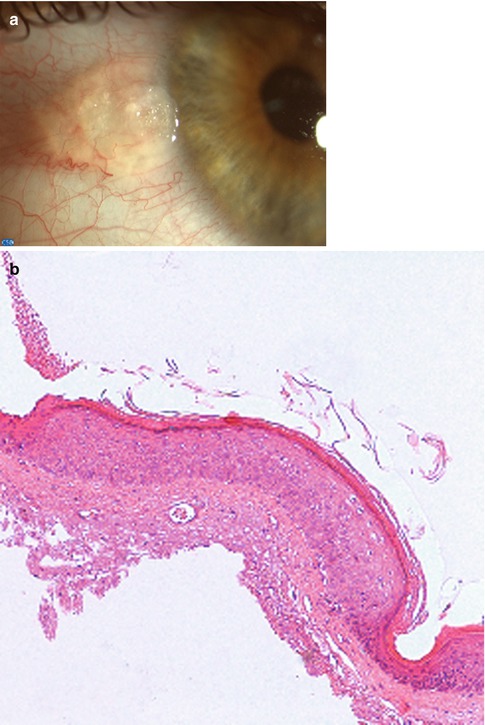
Fig. 2.11
Actinic keratosis. (a) Appearance of a limbal, vascularized, pearly lesion. (b) The conjunctival epithelium is thickened and shows acanthosis, parakeratosis, and mild dysplasia. Actinic elastosis is seen in the substantia propria
2.7.1.2 Ocular Surface Squamous Neoplasia (OSSN)
Definition
OSSN represents a spectrum of disease ranging from mild dysplasia to invasive squamous cell carcinoma (SCC).
Epidemiology
The incidence of conjunctival squamous cell carcinoma varies geographically, declining with greater distances from the equator, with Uganda having 1.2 cases per 100,000 persons per year, compared to the USA with an incidence of 0.03 per 100,000 persons per year.
Etiology
Traditionally, OSSN has been associated with the risk factors of male gender, old age, and ultraviolet light exposure and regarded as a relatively low-grade malignancy with uncommon intraocular invasion and metastasis. In the last decades, the increased knowledge about the oncogenic influence of viruses, particularly HPV types 16 and 18 and human immunodeficiency virus (HIV), has led to the investigation of immunosuppression in the oncogenesis of conjunctival squamous cell carcinoma. More recently, HIV positivity was confirmed to be a highly significant risk factor for the development of SCC. In areas where HIV is endemic, SCC has become increasingly common and is presenting in much younger patients than before, with a dramatic shift toward female predominance.
Localization and Clinical Features
Mild dysplasia, intraepithelial neoplasia, and invasive squamous cell carcinoma are difficult to distinguish on clinical appearances alone. The lesions are described as being slightly elevated, variably shaped, relatively sharply demarcated from the surrounding normal tissues, accompanied by feeding blood vessels and colored from pearly gray to reddish gray depending on the vascularity of the tumor. They most commonly straddle the nasal or temporal limbus between the palpebral fissures [49, 50].
Conjunctival intraepithelial neoplasia (CIN) identifies the precancerous form of OSSN. It typically involves the limbus; is slightly elevated, with well-defined margins; and may be papilliform, leukoplakic, or gelatinous with characteristic tufts of blood vessels. Corneal involvement may be evident as contiguous areas of grayish epithelial thickening. Often the corneal neoplasia has a characteristic fimbriated margin with isolated clusters of gray spots (Fig. 2.12). Most lesions are unilateral.
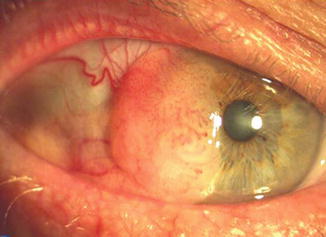
Fig. 2.12
Conjunctival intraepithelial neoplasia. There is a fleshy mass with characteristic tufts of blood vessels, located at the limbus and invading the cornea
Histopathology
Microscopic examination reveals the abrupt transition between the normal uninvolved epithelium and the epithelial neoplasia. Histologically, CIN connotes a partial-thickness to full-thickness intraepithelial neoplasia. Both lesions, however, are restrained by an intact basement membrane without invasion of the underlying substantia propria. The involved epithelium is thickened. The lesion may show mild, moderate, or severe dysplasia. The neoplastic cells characterized by hyperchromatic atypical nuclei and scant cytoplasm may be confined to the basal third of the epithelium (mild dysplasia), or the whole epithelium may be replaced by atypical, pleomorphic cells, with loss of cell polarity and mitotic figures (carcinoma in situ) (Fig. 2.13). Most CINs do not progress to invasive squamous carcinoma.
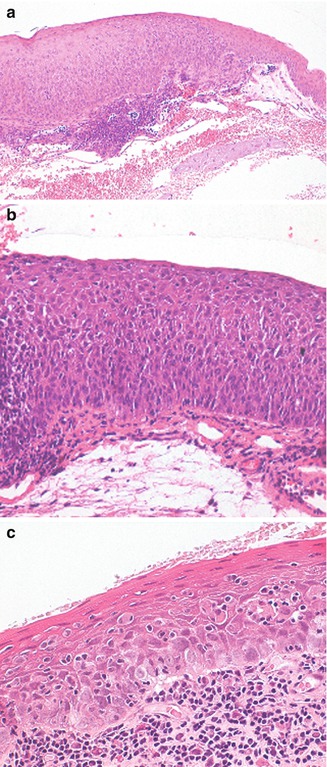
Fig. 2.13
Histopathological characteristics in conjunctival intraepithelial neoplasia (CIN). (a) Moderate dysplasia in CIN. The epithelium is thickened and largely replaced by atypical cells with hyperchromic and pleomorphic nuclei and scanty cytoplasm. Mitotic figures are restricted to the basal two-thirds of the epithelium. A slight surface differentiation is present and the basal membrane is intact. (b) Carcinoma in situ. The thickened epithelium has been completely replaced by spindle-shaped cells with severe nuclear and cytoplasmic atypia, loss of polarity, numerous mitoses, and absence of surface differentiation. Basement membrane is intact. (c) Carcinoma in situ with pagetoid features
Prognosis
Despite its low virulence, CIN is difficult to cure. Recurrence rate may be as high as 50 % if the lesion is incompletely excised, and actually, positive pathologic margins are the strongest predictors of clinical recurrence. Mitomycin C, 5-fluorouracil, and interferon alpha 2b are used as the first treatment modality or as adjuvant therapy for incompletely excised or recurrent lesions [1, 43, 44].
Squamous cell carcinoma may arise de novo or from a preexisting CIN. Lesion can be papillary in configuration with many blood vessels and forms an exophytic mass, leukoplakic with a whitened keratinized plaque, or gelatinous and fleshy. The tumor, located within the interpalpebral fissure, often extends onto the cornea but is not strongly adherent.
Histopathology
In addition to the epithelial changes of CIN, there is invasion by the malignant squamous epithelial cells through the epithelial basement membrane deep into the subepithelial tissue or even into adjacent structures. These lesions are usually well-differentiated carcinomas (Fig. 2.14). The substantia propria exhibits inflammatory cell infiltrate and contains atypical epithelial cells greatly variable in size and configuration, and depending on the degree of differentiation, the tumor may show hyperplastic and hyperchromatic cells, keratinized cells, concentric collections of keratinized cells (keratin pearls), loss of cellular cohesiveness, and atypical mitotic figures (Fig. 2.15). Conjunctival squamous cell carcinoma intensely expresses immunoreactivity for the tyrosine kinase EGF receptor. CIN and CSCC are associated with preferential expression of p63 in the immature dysplastic epithelial cells.
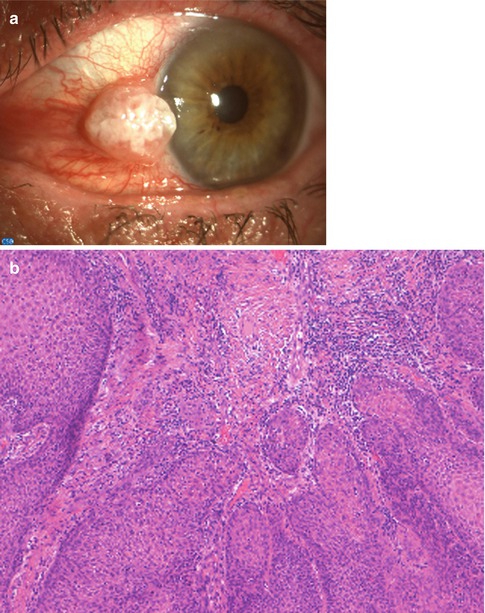
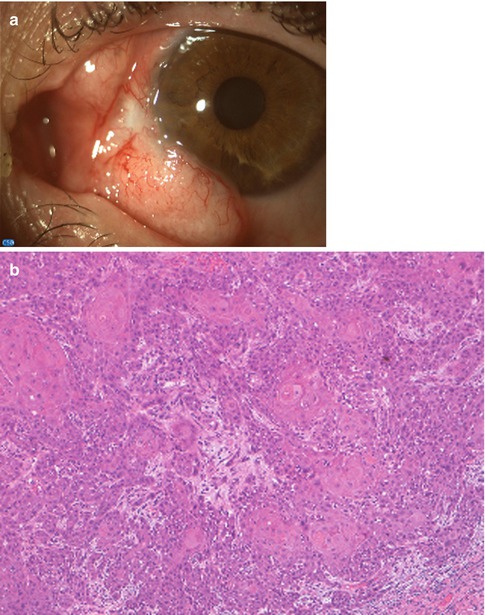
Fig. 2.14
Squamous cell carcinoma. (a) An exophytic, leukoplakic mass is present at the limbus. (b) Light microscopical examination reveals a well-differentiated carcinoma. The conjunctival stroma is infiltrated by nests of neoplastic squamous cells with large eosinophilic cytoplasm, intercellular bridges, and keratin pearls
Fig. 2.15
Squamous cell carcinoma with superficial invasion. (a) There is a large, bilobate, vascularized mass of the bulbar conjunctiva. (b) Histopathologically, the basal membrane is interrupted. The epithelial cells display keratinization. At the edge of the infiltrating lobules, the cells have a more atypical appearance
Spindle cell carcinoma is a rare variant of squamous cell carcinoma that may behave in a rather aggressive fashion. It is characterized by pleomorphic spindle cells with variably shaped nuclei, in comparison to the uniformity of the more common spindle cell, and frequent mitotic figures. Positive staining with cytokeratin, epithelial membrane antigen markers, and p63 is helpful in differentiating the variant from other spindle cell tumors such as amelanotic melanoma, malignant schwannoma, fibrosarcoma, leiomyosarcoma, and malignant fibrous histiocytoma (Fig. 2.16) [51].
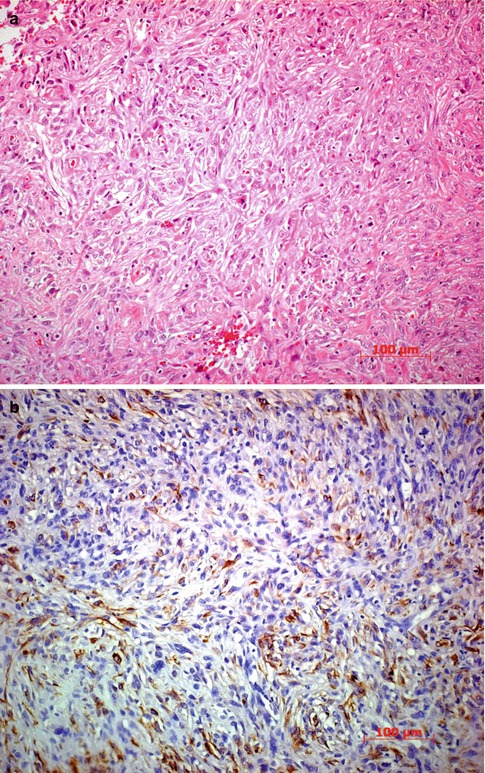
Fig. 2.16
Spindle cell carcinoma. (a) Histopathologically, the tumor cells grow in the conjunctival stroma beneath an intact epithelium. HE stain. (b) Immunohistochemical stain for cytokeratins 10, 17, and 18 is positive in most of the spindle tumor cells. The spindle cell type is more prominent in this microphotograph
Mucoepidermoid carcinoma contains cuboidal mucus-secreting cells with intracytoplasmic droplets, intermixed with epidermoid cells. Cells are arranged in nests and cords with secreted pools of mucin in the extracellular spaces. Mucinous secretion is detectable with the aid of special stains such as mucicarmine, alcian blue, and periodic acid–Schiff. A third type of cell, called intermediate or basal cell, may also be found. These tumors are uncommon in the conjunctiva and usually arise in elderly individuals in the seventh decade of life. They appear to be aggressive locally and tend to recur rapidly after excision [52, 53].
Basal cell carcinoma may rarely occur in the actinically exposed conjunctiva. Histologic examination discloses invasive epithelial lobules with a palisading outer border and peripheral cleft [1, 43, 54, 55].
Prognosis
Carcinoma of the ocular surface is generally regarded as low-grade malignancy. However, intraocular invasion has been reported in 11 %; corneal and/or scleral invasion was found in 30 %, and orbital invasion was noted in 15 % of patients (Fig. 2.17) [56]. Complete resection is the predominant treatment modality for OSSN, and the ocular surface is frequently reconstructed with amniotic membrane. Cryotherapy and/or topical chemotherapy (mitomycin C, 5-fluorouracil, interferon alpha-2b) may be considered as adjunctive therapies. Orbital exenteration may be required for extensive tumor involvement of orbital soft tissues. Clinical and pathologic staging is recommended, according to American Joint Committee on Cancer, using the TNM classification [57]. Histologic grade includes five categories: GX, grade cannot be assessed; G1, well differentiated; G2, moderately differentiated; G3, poorly differentiated; and G4, undifferentiated.