The Eye and Renal Diseases
Michaella Goldstein
Gad Heilweil
In 1836, the distinguished English physician Richard Bright was the first to associate renal disease with blindness. Two decades later, the German physician and pharmacologist Matthias Liebreich described fundus changes in uremic patients. The condition was eventually labeled “Bright disease” (albuminuric retinitis), and by the end of the 19th century, it was no longer considered to be a separate entity but a manifestation of hypertension in uremic patients. Contemporary treatment for hypertension has felicitously led to diminishing numbers of patients who present with severe hypertensive changes in the fundus. At the same time, however, the prolonged survival rate of patients undergoing dialysis and renal transplantation has resulted in greater numbers of patients with drug-induced side effects, as well as with complications of dialysis or renal transplant, some of which directly or indirectly affect vision. This review will present the pathological entities in which kidney disease and ocular injury can be linked in order to raise the level of awareness of the practicing physician to this association.
Diabetes and hypertension are by far the diseases most responsible for visual disturbances associated with renal pathology. This chapter will cover the most of the other conditions that have been found concomitant with renal disease.
Ocular Complications of Renal Failure, Hemodialysis, and Renal Transplantation
Patients undergoing hemodialysis are subject to the affect of both uremia and the dialysis procedure on multiple organ systems. The manifestation of this injury in the eye can range from mild changes in intraocular pressure for which expectant follow-up is sufficient to potentially devastating retinal detachment with severe visual loss that requires emergent surgery.
Intraocular Pressure Changes
The aqueous humor is formed by the non pigmented epithelium of the ciliary body due to active transport of ions and nutrients from the circulation. Another portion of the aqueous humor is derived by ultrafiltration of interstitial fluids. There appear to be two opposing intradialytic forces that alter intraocular pressure (IOP). The ultrafiltration during hemodialysis draws fluid out of tissues and can lead to a decrease in IOP toward the end of the dialysis session.1 On the other hand, removal of uremic toxins and other solutes during dialysis can lower serum osmolarity more rapidly than ocular osmolarity, causing an osmolar gradient, which leads to water movement into the aqueous humor and an increase in IOP.2 It has been shown that elevated IOP in dialysis patients correlates with high post – dialysis urea rebound and rapid solute removal.3 The eye usually seems to be able to compensate for these fluid alterations so that an increase in IOP mostly occurs in patients with impaired outflow.4 The risk of IOP fluctuations can be reduced by increasing the time of the dialysis session.
Corneoconjunctival Abnormalities
Dialysis is aimed at controlling the calcium phosphate product, which means lowering systemic calcifications. In patients with renal failure, hypercalcemia may nevertheless lead to soft tissue calcification, which can often be detected in the interpalpebral cornea and conjunctiva. The corneal calcification that is deposited beneath the cornea epithelium produces band keratopathy and epithelial erosions, causing decreased visual acuity, red eye, and severe pain. Deposits can be usually removed by excimer phototherapeutic keratectomy or superficial keratectomy with or without chelating with calcium and ethylenediamine tetra-acetic acid (EDTA).5
Cataract
It is unclear whether cataracts are more common in patients with end-stage renal disease, but early cataract formation in young patients is well recognized and may be attributed to underlying diseases, such as diabetes or prolonged use of corticosteroids.
Retinal Abnormalities
Serous retinal detachment mimicking central serous chorioretinopathy may be seen in patients undergoing hemodialysis6 or receiving prolonged treatment with steroids after renal transplantation.7 Patients with renal insufficiency have impaired fluid and electrolyte balance as well as dysfunction of the overlying retinal pigment epithelium, leading to focal subretinal edema and exudative retinal detachment (Fig. 1).8 Bilateral bullous retinal detachment, multiple retinal pigment epithelial detachment, and yellow subretinal exudates beneath the sensory retinal detachment may be seen in severe forms. Retinal hemorrhages are of particular concern in dialysis patients on heparin, many of whom have proliferative diabetic retinopathy. In one study on diabetic patients, however, loss of vision was found to be independent of dialysis modality, suggesting that heparin use was safe in this context.9
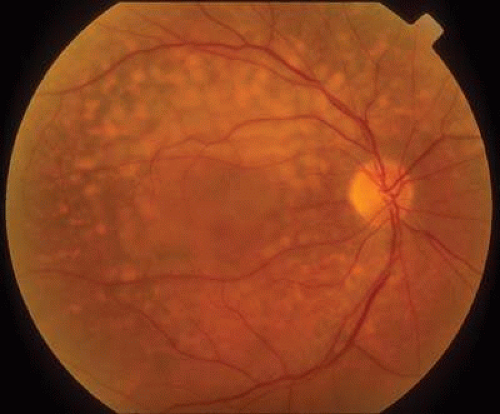 Fig. 1. Color fundus picture of a 73-year-old man with end-stage renal failure, demonstrating diffuse yellow drusen-like deposits, and neurosensory elevation of the posterior pole. |
Optic Neuropathy
Ocular Infection
Renal transplant recipients are immunocompromised, thus making them more prone to develop ocular and periocular infections. Viral retinitis, usually caused by cytomegalovirus, is the most severe ocular complication. Valganciclovir has been shown to be effective in both preventing and treating cytomegalovirus retinitis in renal transplant patients.12 Bacterial and fungal organisms have also been associated with endophthalmitis in renal transplant patients.
Neoplastic Complication after Renal Transplantation
There have been several reports of ocular neoplastic involvement in renal transplant patients. Squamous cell carcinoma of the eyelid and the conjunctiva has been reported in renal transplant patients receiving long-term cyclosporin therapy.13,14 Brown tumor of the orbit is very rare and was observed in several patients with chronic renal failure and secondary hyperparathyroidism; it is a focal bony lesion caused by increased osteoclastic activity with bone resorption and trabecular fibrosis.15
Oculorenal Syndromes
Combined disorders of the eyes and kidneys may result from a metabolic or a developmental defect, vascular or autoimmune disease, infection, neoplasia, or toxicity, comprising a large group of inherited and noninherited malformations and multisystemic diseases. These disorders are classified in Table 1. The classic and most clinically important are discussed in detail.
TABLE 1. Etiologic Classification of the Oculorenal Syndromes | |
|---|---|
|
Chromosomal Abnormality Syndromes
Aniridia–Wilms Tumor
Wilms tumor is the most common malignant renal tumor in both children and adolescents.16 The tumor is derived from the developing renal blastema (embryonic renal precursor) that has failed to differentiate normally.17 The majority of Wilms tumor cases are diagnosed by the age of 6 years.18 Wilms tumor follows an autosomal dominant inheritance pattern with incomplete penetrance. Some patients have chromosomal abnormalities that include cytogenetic deletions on the short arm of chromosome 11, involving 11p13.19 The WT1 gene was found to be a tumor suppressor gene responsible for a few cases of congenital absence of all but the root of the iris (aniridia).17 Recent studies have shown the WT1 gene has a role in retinal formation and especially on ganglion cell development.20
Wilms tumor is associated with congenital ocular abnormalities including aniridia (Fig. 2), both sporadic and in association with WAGR syndrome (Wilms tumor, sporadic aniridia, genitourinary malformations, and mental retardation), and less commonly optic nerve hypoplasia, cataract, and glaucoma.18 Ocular abnormalities are thought to result from disruption of the aniridia gene PAX6 that lies on the distal end of the chromosome.21
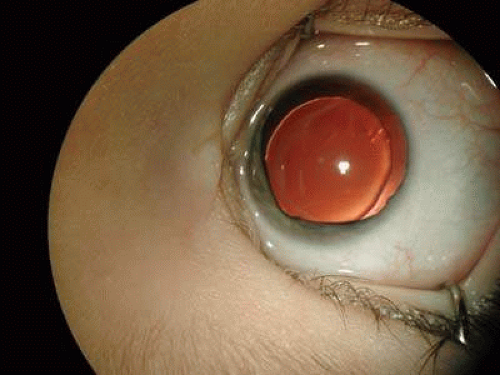 Fig. 2. Color picture of a 3-year-old girl with WAGR syndrome, presenting at the age of 3 months with bilateral aniridia, who had consequently developed Wilms tumor at the age of 5. (Courtesy of Chaim Stolovitch, M.D., Sackler School of Medicine, Tel-Aviv University.) |
In the absence of a specific marker for Wilms tumor, the role of the ophthalmologist is crucial in recognizing the disease. The detection of aniridia in young children should alert for Wilms tumor.
Cat-eye Syndrome
Cat-eye syndrome is a very rare disorder, derived from a defect in chromosome 22. Typical cat-eye syndrome includes iris coloboma and anal atresia. Other ocular features are choroidal or optic nerve coloboma and microphthalmia.22 Variable renal abnormalities such as unilateral absence, unilateral or bilateral hypoplasia, and cystic dysplasia have also been reported.23 Other systemic manifestations include preauricular skin tags and/or pits (which are probably the most consistent feature), congenital heart defect, and usually low-normal intelligence.
Autosomal Dominant Syndromes
Acro-renal-ocular Syndrome
Duane-radial ray syndrome/Okihiro (DRRS) is an autosomal dominant condition characterized by radial ray defects and Duane anomaly (limitation of abduction with narrowing of the palpebral fissure and globe retraction on adduction).24 This syndrome may include other abnormalities such as anal, renal, cardiac, ear, and foot malformations and hearing loss. Acro-renal-ocular syndrome differs from DRRS by the presence of structural eye anomalies such as microcornea and uveal and optic nerve colobomas.25 Both syndromes are the result of a mutation in the SALL4 gene. The SALL4 gene product is a zinc finger protein that is thought to act as a transcription factor.26 The radial ray abnormalities of the hand vary in expression from mild thenar hypoplasia or inability to flex the interphalangeal joint of the thumb to hypoplastic thumbs or prominent upper limb abnormalities. The urinary tract anomalies include unilateral renal agenesis, renal ectopia, malrotation, and bilateral renal hypoplasia.
Nail-Patella Syndrome
Nail-patella syndrome (hereditary osteo-onychodysplasia) is a rare autosomal dominant disorder of nails, skeleton, and kidney. The nail-patella gene has been mapped to chromosome 9 (9q34), and it is closely linked to the genes coding for ABO blood groups and for adenylate kinase.27 The nails are hypoplastic or absent, particularly affecting the thumb and index finger. Characteristic skeletal abnormalities include small or absent patellae, iliac horns, and abnormality of the elbows interfering with pronation and supination. Recurrent subluxations of knees and elbows, flexion deformities of the elbows, and Madelung deformity with volar position of the wrists are often present in patients with nail-patella syndrome but are rarely incapacitating. Clinical renal involvement occurs in about 30% to 40% of cases, and proteinuria that is sometimes associated with microhematuria is the most frequent feature.28 End-stage renal failure may occur, and nephropathy may be the most serious complication in some patients. Collagen fibrils in the thickened basement membrane are demonstrated by electron microscopy.29 Typical renal lesions are focal glomerular basement membrane thickening with subendothelial fibrillar electron-dense deposits and moth-eaten appearance of the glomerular basement membrane. A cloverleaf dark pigmentation of the central area of the iris with scalloped iris collarette (Lester sign of the iris) is a peculiar finding in some patients. Strabismus, keratoconus, microcornea, sclerocornea, microphakia, and cataracts have also been described in patients with this syndrome.28
Papillorenal Syndrome
Papillorenal syndrome was defined by Bron et al.30 as a dominantly inherited disorder with a bilateral dysplasia of the optic discs associated with a severe form of glomerulonephritis that may lead to renal failure. It is probably a primary dysgenesis that causes vascular abnormalities affecting the eye, kidney, and urinary tract, leading to hypoplasia of these structures. The pathogenesis may be a mutation in the PAX2 genes, which are developmental control genes that encode nuclear transcription factors. Characteristic findings consist of an absence or attenuation of the central retinal vessels within the optic nerve and the presence of multiple compensatory cilioretinal vessels.31 Supranasal visual field defects can be seen. The ophthalmologist should consider screening for renal disease in all patients with multiple cilioretinal vessels suspected of having papillorenal syndrome.
Renal-Coloboma Syndrome
Renal-coloboma syndrome is an autosomal dominant disorder that is characterized by bilateral optic nerve coloboma or dysplasia and renal hypoplasia. Additional congenital defects, such as vesicoureteral reflux, and auditory and central nervous system (CNS) anomalies, may occur with variable penetrance.32 The pathogenesis is a mutation in a PAX2 gene, a developmental control gene on chromosome 10q24.3–25.1.33,34 The colobomatous defect in the eye involves the optic nerve or posterior pole. The kidneys are usually small, with reduction of both the renal pelvis and the renal cortex. The renal disease is usually progressive and frequently necessitates renal transplantation after chronic renal failure.32,35 Pathologic examination reveals cortical thinning and low numbers of glomeruli and collecting ducts.
Von Hippel–Lindau Disease
Von Hippel–Lindau disease (VHL) is an autosomal dominant neoplasia syndrome predisposing to ocular and CNS hemangioblastomas, renal cell carcinoma, and pheochromocytoma, as well as cysts in various organs, all resulting from a germline mutation in the VHL gene. VHL is a tumor suppressor gene on the short arm of chromosome 3 (3p 25–26).36 Tumor formation is initiated when both VHL alleles are inactivated.37 The incidence of VHL is 1:36,000 live births, and there is an almost 90% penetrance until the age of 65 years. The cumulative risk of a patient with VHL disease to develop retinal hemangioma, cerebral hemangioblastoma, and renal cell carcinoma at age 30 years are 44%, 38%, and 5%, respectively, rising to 84%, 70%, and 69%, respectively, at the age of 60 years.38 Advances in genetic testing include qualitative and quantitative Southern blotting, which, together with DNA sequence analysis, increase the detection rate of DNA mutation in peripheral blood leukocytes from 75% to nearly 100%.39 Renal cell carcinoma is the most frequent cause of death, followed by cerebellar hemangioblastoma.40 Renal lesions are often multiple and bilateral. Contrast-enhanced abdominal computed tomography is the standard for renal involvement detection and is useful for early diagnosis; however, in some cases the diagnosis is accomplished only with the appearance of hematuria, flank pain, or flank mass.41
Hemangioblastoma of the CNS together with retinal hemangiomas are the most common tumors in VHL, affecting 60% to 80% of all patients.42 The CNS hemangioblastomas are benign tumors and can arise anywhere along the craniospinal axis but mainly in the spinal cord and cerebellum. The retinal hemangioblastomas arise in the periphery or near the optic nerve or both. They are often multifocal and bilateral, and the mean age of patients at presentation is 25 years. Both peripheral and central angiomas can cause visual symptoms due to increased vascular permeability and the development of hard exudates in the macula, and this can cause exudative and tractional retinal detachment. Screening should include at least a once-yearly indirect ophthalmoscopy and preferably fluorescein angiography for early detection of small angiomas. Small tumors respond well to laser photocoagulation, and if they are adjacent to the optic nerve, they may respond well to photodynamic therapy, while larger ones at the periphery will require cryocoagulation.43,44
Autosomal Recessive Syndromes
Bardet-Biedl Syndrome and Related Disorders
Bardet-Biedl syndrome is an autosomal recessive disease that causes severe visual impairment due to retinitis pigmentosa (rod-cone dystrophy). The disorder is characterized by retinitis pigmentosa, obesity, polydactyly, renal abnormalities, hypogonadism and variable degrees of mental retardation.45 Six BBS genes have been characterized, and eight different loci of mutations were identified on various chromosomes.46,47 Variability in the systemic phenotypes has been described, but retinal dystrophy is an essential feature of the syndrome.48
The cone-rod dystrophy usually appears at the end of the first decade of life and causes central and peripheral visual loss by the second or third decade, with the majority of affected individuals being legally blind by that time. High myopia is common, and the retinopathy is usually without the typical bone spicule – like pigmentation, attenuated retinal vessels (often with significant surface retinal wrinkling), and pale optic disc. The electroretinogram is a sensitive detector of retinopathy in patients with Bardet-Biedl syndrome and may allow identification of the photoreceptor cell degeneration in young children with normal fundus appearance.49,50
In addition to classic Bardet-Biedl syndrome, several variants and related disorders with or without ocular abnormalities have been described. They include Laurence-Moon syndrome and Biemond and Alström syndromes. Patients with Laurence-Moon syndrome share most of the features of Bardet-Biedl patients, but they are spastic and do not have polydactyly. Patients with Biemond II syndrome have iris coloboma and lack pigmentary retinopathy. Alström syndrome is characterized by retinal dystrophy, obesity, cardiomyopathy, neurosensory hearing loss, and insulin-resistant diabetes mellitus.51 The gene for Alström syndrome (ALMS1) has been localized to human chromosome 2p13.
Cockayne Syndrome
Cockayne syndrome is a DNA repair disorder.52 It is a progressive neurological disorder characterized in infancy by growth failure, deficient neurological development, progressive retinal degeneration, and sensitivity to sunlight. One of the hallmarks of Cockayne syndrome is pigmentary degeneration of the retina, which occurs in more than 60% of reported cases.53 electroretinogram responses in these patients shows variable degrees of reduction of scotopic and photopic responses. Renal complications develop in about 10% of patients with Cockayne syndrome and can progress to renal failure or hypertensive crisis secondary to the renal damage. Histologic findings demonstrate thickening of the glomeruli basement membrane.
Nephropathic Cystinosis
Cystinosis is an autosomal recessive disease caused by intracellular cystine accumulation due to a defect in the lysosomal cystine carrier leading to accumulation and deposition of cystine crystals in many body tissues, including the kidneys and the eyes. The juvenile form of nephropathic cystinosis is the most common and severe variant. The symptoms are similar but usually milder in late-onset or adolescent cystinosis. Benign or adult cystinosis is a rare nonnephropathic variant. All forms of cystinosis result from a mutation in the CTNS gene (cystinosis gene) located on chromosome 17p13.54 Numerous mutations in the CTNS gene were found in the three variants of cystinosis.55 Heterozygotes can be detected by measuring the cystine content of polymorphonuclear leukocytes, and prenatal diagnosis of nephropathic cystinosis can be made by using cultured amniocytes or chorionic villi.
The systemic features of nephropathic cystinosis usually present in the first year of life with failure to thrive and dehydration due to proximal renal tubular dysfunction. Rickets and corneal crystals often appear by 1 to 2 years of age. The typical phenotype of patients with nephropathic cystinosis includes short stature and light complexion. As a result of progressive renal failure, end-stage renal disease generally occurs by the end of the first decade of life, necessitating dialysis or renal transplantation. In young children, growth can be enhanced and renal deterioration can be delayed or prevented by cysteamine, a cystine-depleting agent.56 Since the advent of renal transplantation, many cystinotic patients live to adulthood and become susceptible to new complications caused by long-standing cystine accumulation in nonrenal organs. Cerebral atrophy can be found on computed tomographic scans of older patients, and a large spectrum of neurologic features has been described, including nonabsorptive hydrocephalus and a peculiar form of encephalopathy, leading to a “pseudobulbar” state.57 Hypohidrosis, hypothyroidism, late sexual maturation, insulin-dependent diabetes, liver enlargement with portal hypertension, and severe epistaxis have been observed in several patients who survived into their second and third decades.56,58
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


