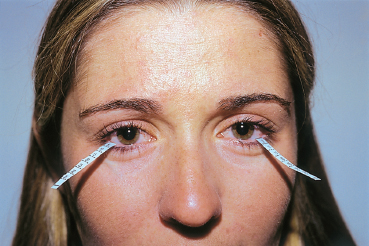
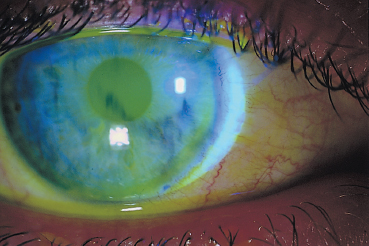
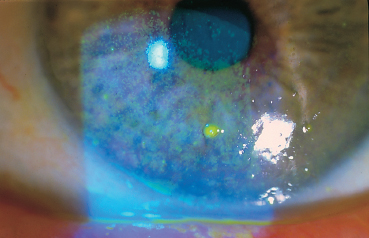
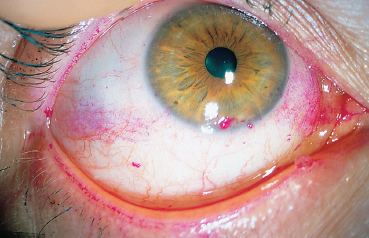
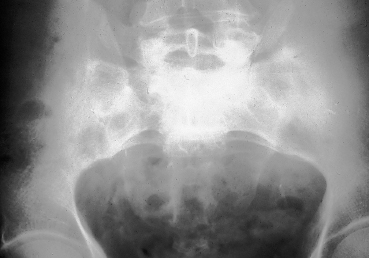
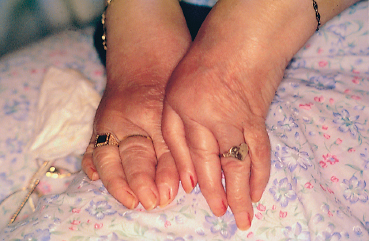
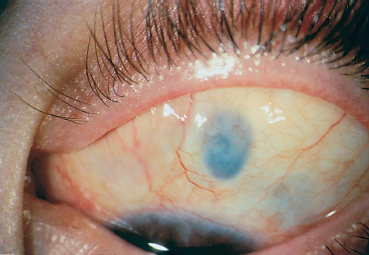
Chapter 9 Systemic Diseases and Conjunctivitis Fig. A Schirmer I test Fig. B Tear film break-up time: biomicroscopic examination with blue light illumination shows “dark spots” in the precorneal tear film Fig. C Epithelial punctate keratitis and filamentary keratitis Fig. D Rose bengal staining in the interpalpebral exposure zone Fig. E Lissamine green staining Fig. F Lymphocytic sialadenitis in a patient with Sjögren’s syndrome. Periductal lymphocytic infiltration with migration of some lymphocytes into the epithelium. The ducts are mildly ectatic. The remainder of the glandular parenchyma contains a dense infiltrate mainly composed of plasma cells (accessory salivary gland biopsy, HES staining, x250) (Courtesy of Professor B. Kantelip, Besançon) Usually, the conjunctivitis itself has nothing particularly specific and is rarely a key element in the diagnosis of the underlying condition. However, the conjunctivitis must be diagnosed, and an association with inflammation of adjacent tissues such as the episclera, sclera, corneal limbus, or peripheral cornea can be more informative. Investigators should not forget that the conjunctiva is a readily accessible source of tissue samples and can provide useful diagnostic information on an underlying systemic disease, as well as its pathogenesis and therapeutic indications. As an exhaustive description of the different systemic diseases that may be accompanied by inflammation of the conjunctiva is beyond the scope of this chapter, the reader is invited to consult reference works for further details. Ocular manifestations of the posterior segment will not be dealt with here. When a patient is known to have a systemic disease, it may appear simple and logical to attribute any conjunctival involvement to it. In contrast, conjunctivitis is usually nonspecific and, when it is the presenting sign, its clinical characteristics alone are rarely sufficient to diagnose the causal systemic disease. However, conjunctival manifestations rarely remain isolated, but are rather part of an ensemble of presumptive factors leading to diagnosis of the primary disease. Conjunctival lesions are readily accessible to direct examination, and sometimes offer the possibility of exploring the underlying systemic disease. The pathogenesis of the conjunctival involvement is usually identical to that of the underlying disease; like other sites of disease involvement, conjunctival manifestations can be an index of progression and a prognostic indicator. Thus, the onset of conjunctival complications may reflect a possible aggravation of the systemic disease, which is otherwise silent. We have chosen to classify systemic diseases by the main organ systems and pathogenic mechanisms. This is, of course, an arbitrary choice, as most of the diseases mentioned below affect several organs. Similarly, although there is a separate section on “Immunological Diseases,” it should be borne in mind that immunological disturbances are involved in many other diseases classified under different headings. To help the reader, we have tried, when possible, to list the signs with the best specificity and sensitivity to confirm the clinical impression. At the end of this chapter, we have included the different conjunctival manifestations in the clinical contexts that are usually encountered in practice. Locomotor diseases are the systemic diseases most often associated with conjunctival involvement, in which the latter can reveal the former. We will not deal here with conjunctival involvement during autoimmune bullous diseases, allergies, or rosacea, which are covered in other chapters. Review of Systemic Diseases Locomotor Diseases Reiter’s Syndrome and HLA-B27-Associated Reactive Arthritis Reiter’s syndrome belongs to the poorly defined nosological entity designated “reactive arthritis.” The latter is defined as arthritis due to microbial infection in which no pathogens are detected at the site of the involved joint by means of conventional bacteriological techniques.1 Ankylosing spondylarthritis differs from Reiter’s syndrome in that it is not accompanied by involvement of the mucosae (conjunctiva, mouth, urethra) or skin. Reiter’s syndrome is characterized by the classical combination of arthritis, conjunctivitis, and urethritis. It mainly affects young men (Fig. 9.1). It is often venereal in origin; Chlamydia trachomatis and Ureaplasma urealytica are frequently incriminated (see Chapter 7). Conjunctivitis is present in 58% of cases and is often bilateral. It is usually moderate and associated with mu-copurulent secretions. The inflammation generally resolves in 7-10 days and microbiological investigations are negative.2 Patients rarely consult a specialist because the symptoms are mild, even if conjunctivitis is often present at onset. It can, however, take on a hyperacute form, with abundant secretions and subconjunctival hemorrhage. Scleritis and episcleritis are rare, as is corneal and uveal involvement. The histocompati-bility antigen HLA-B27 is present in 80% of Caucasian patients with Reiter’s syndrome.3 Reactive arthritis with an intestinal portal of entry can also occur during shigellosis, yersiniosis, salmonellosis, and Campylobacter pylori infection, the HLA B27 antigen being presented to immunocompetent cells together with bacterial antigens. Diagnostic laboratory test: HLA-B27 antigen. Relapsing Polychondritis Relapsing polychondritis is a rare and severe systemic disease of unknown origin, characterized by recurrent inflammation of articular and nonarticular cartilage and tissues rich in pro-teoglycans. Diagnostic criteria include the following: auricular chondritis, cochlear or vestibular damage, chondritis of nasal cartilage, tracheal and laryngeal chondritis, nonerosive inflammatory polyarthritis, and ocular inflammation (Fig. 9.2). Fig. 9.1 Sacroiliitis in a patient with Reiter’s syndrome (Courtesy of Professor C. Tavernier, Dijon) Ocular involvement is the initial manifestation in 20% of cases and is found in 60% of patients during the course of the disease. All ocular structures can be affected.4 Palpebral inflammation is sometimes so intense that it resembles orbital cellulitis. Ptosis and chemosis can also mimic an inflammatory pseudotumor. Episcleritis, scleritis and iritis are the most common ocular manifestations of relapsing polychondritis.5,5a Ker-atoconjunctivitis sicca is generally moderate unless it is associated with Sjögren’s syndrome.6 Other ocular manifestations include keratitis, chorioretinitis, and optic neuritis. Rheumatoid Arthritis Rheumatoid arthritis (RA) is a chronic inflammatory multisystem disease classically causing symmetric polyarthropathy (Fig. 9.3). Its diagnosis is based on an ensemble of well-defined clinical criteria (Table 9.1)7 and the presence of nonspecific but highly evocative rheumatoid factors (anti-IgM antibodies) in 70% of cases (compared to 1-5% in the general population). The pathogenesis of the ocular manifestations is identical to that of the extraocular signs: autoimmunity causes tissular inflammatory infiltration, sometimes associated with necrosis and vasculitis which is mainly responsible for neurooph-thalmological complications. Onset usually occurs between the fourth and fifth decades. Approximately 80 % of cases occur in women.8 Extraarticular involvement is common, and is more frequent in patients who are rheumatoid factor-positive and/or have severe joint involvement. Fig. 9.2 Relapsing polychondritis: saddle nose deformity due to nasal chondritis Fig. 9.3 Rheumatoid arthritis: typical hand deformity Fig. 9.4 Rheumatoid arthritis: scleromalacia perforans Table 9.1 American Rheumatism Association (ARA) (1988) criteria for the diagnosis of rhematoid arthritis (RA)7
| 1—Morning stiffness 2—Arthritis in 3 or more joints 3—Arthritis of hand joints 4—Symmetric polyarthritis 5—Rheumatoid nodules 6—Serum rheumatoid factor 7—Radiographic changes (More than 4 criteria necessary for diagnosis. Criteria 1-4 present for at least 6 weeks) |
Keratoconjunctivitis sicca, found in 10-25% of patients, is the most frequent form of ocular involvement in RA. It can cause epithelial punctate keratitis, the gravity and outcome of which are independent of the severity of RA. Keratoconjunctivitis sicca associated with xerostomia can be part of secondary Sjögren’s syndrome. Sjögren’s syndrome is found in 25% of patients with RA, but 60% of patients with dry mouth and eyes have or will develop a system disease.9
The other anterior ocular manifestations of RA are epis-cleritis, scleritis (sometimes scleromalacia perforans) (Fig. 9.4), and peripheral ulcerative keratitis.
Diagnostic laboratory test: serum rheumatoid factor.
Connective Tissue Diseases
Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is a multisystem disease with a strong female predominance (male/female sex ratio = 1/9) that tends to begin in the second or third decade. It is principally a musculoskeletal disease, diagnosed on the basis of an ensemble of clinical and laboratory arguments (Table 9.2). The characteristic malar “butterfly” rash is present in 30% of cases (Fig. 9.5). Cutaneous involvement is highly variable, consisting of erythematous or atrophic lesions; biopsy of these lesions sometimes establishes the diagnosis by showing, in direct immunofluorescence studies, the presence of a lupus band (Fig. 9.6). This corresponds to immunoglobulin deposits at the basement membrane zone (BMZ). Joint involvement is frequent (50% of patients), with migrating arthralgia accompanied by myalgia. Pulmonary, cardiac, and renal complications determine the prognosis. The presence of native antinu-clear and anti-DNA antibodies is highly evocative. Other antibodies such as those to SS-A/Ro and SS-B/La, and RNP, can be found in patients with associated secondary Sjögren’s syndrome and Raynaud’s phenomenon, respectively.
Ocular involvement is fairly rare. Keratitis is present in 5.8% of patients. Conjunctival manifestations are mainly associated with secondary Sjögren’s syndrome, which is often mild. In rare cases it can consist of fibrous conjunctivitis11 (Figs. 9.7 and 9.8). Other anterior segment manifestations include phlyc-tenular limbal nodules and peripheral ulcerative keratitis sometimes mimicking Mooren’s ulcer.12
Diagnostic laboratory tests: antinuclear and native anti-DNA antibodies (Ab) (present in two thirds of cases), anti-Sm Ab (present in only 15 % of cases, but almost fully specific). SS-A and SS-B, and RNP autoantigens, respectively, point to associated secondary Sjögren’s syndrome or Raynaud’s phenomenon. Cutaneous biopsy to detect a lupus band.
Progressive Systemic Sclerosis
Progressive systemic sclerosis is a generic term covering a number of diseases characterized by skin fibrosis associated with marked atrophy of the subcutaneous tissue, and visceral complications. The cutaneous involvement (sclerodactyly, acrosclerosis) is the key element from both a diagnostic and a prognostic point of view. The Raynaud’s phenomenon is common. Progressive systemic sclerosis affects many organs, with potentially life-threatening consequences (lungs, heart, digestive tract, and kidneys). The Raynaud’s phenomenon is one of the most common manifestations of progressive systemic sclerosis.13
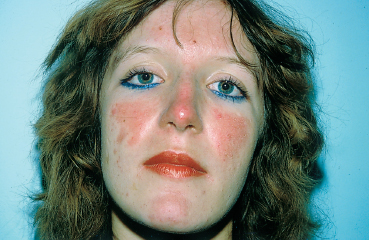
Fig. 9.5 Systemic lupus erythematosus (SLE): malar “butterfly” rash (Courtesy of Dr. S. Dalac, Dijon)
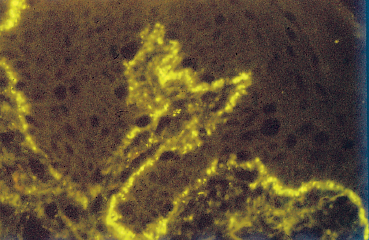
Fig. 9.6 Lupus band (IgG deposits at the cutaneous basement membrane zone) (light microscopy, direct immunofluorescence x 100) (Courtesy of Professor B. Kantelip, Besançon)
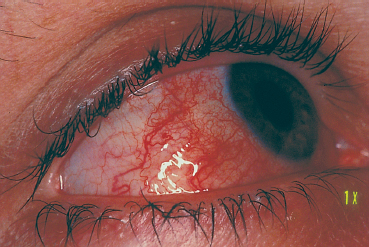
Fig. 9.7 Wegener’s granulomatosis: diffuse scleritis
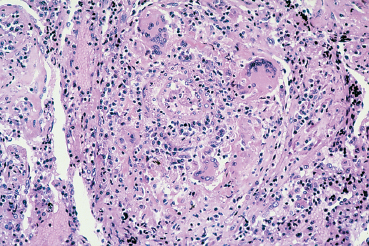
Fig. 9.8 Wegener’s granulomatosis: necrotizing granulomatous vasculitis with presence of giant cells (light microscopy, HES x 400) (Courtesy of Professor B. Kantelip, Besançon)
Progressive systemic sclerosis affects the eyes in 70% of cases. The main symptoms are a foreign-body sensation and intermittent blurred vision. Conjunctival manifestations consist of telangiectasia and sludging of the conjunctival vessels. Fornix foreshortening is also common. Tear insufficiency is the most frequent ocular manifestations of progressive systemic sclerosis,14 affecting 37-79% of patients according to the study. The association of keratoconjunctivitis sicca with xerostomia indicates the presence of secondary Sjögren’s syndrome.15 Conjunctival biopsy can be useful for early diagnosis of progressive systemic sclerosis by showing marked fibrosis associated with sparse infiltrating lymphocytes.
Diagnostic laboratory tests: antinuclear (anticentromere) Ab (present in 90 % of patients at a very high titer), Scl 70 Ab. Capillaroscopy: nailfold capillary changes
Sjögren’s Syndrome (see Focus  8)
8)
Sjögren’s syndrome is a multisystem disease affecting the lacrimal glands, salivary glands, and all the mucous membranes. The glands are destroyed by mononuclear cell infiltration, and are replaced by fibrous tissue. The degree of lympho-proliferation determines the severity of exocrine gland involvement: spread to the viscera provokes the onset of clinical and biological signs similar to those found in connective tissue diseases. Other sites of involvement are responsible for dy-sphagia, hoarseness, cough, renal tubulopathy (distal renal tubular acidosis), atrophic vaginitis, and vasculitis (Raynaud’s phenomenon). There is also a risk of lymphoproliferative disease: patients with Sjögren’s syndrome have a more than 40-fold higher risk of developing salivary nonHodgkin’s lym-phoma than do healthy subjects.16
Table 9.2 American Rheumatism Association (ARA) (1982)10 criteria for the diagnosis of systemic lupus erythematosus (SLE)
| 1—Malar rash 2—Discoid rash 3—Photosensitivity 4—Oral or nasopharyngeal ulcers 5—Nonerosive arthritis affecting at least two peripheral joints 6—Pleurisy, pericarditis, serositis 7—Proteinuria, casts 8—Seizures, psychosis 9—Hemolytic anemia, leukopenia, thrombocytopenia 10—LE cells, native anti-DNA Ab, anti-Sm Ab, false-positive syphilis serology on two occasions 6 months apart 11—Abnormal titer of ANA (anti-nuclear Ab) in the absence of culprit drugs |
Table 9.3 Diagnostic criteria for Sjögren’s syndrome17
1—Ocular involvement: xerophthalmia for more than 3 years, foreign-body sensation, artificial tears more than 3 times per day. 2—Ocular tests: Schirmer test < 5 mm at 5 mins; rose bengal staining score >4 3—Salivary involvement: xerostomia for more than 3 months, recurrent salivary gland swelling, frequent beverages required 4—Salivary gland dysfunction: scintigraphy and/or sialography 5—Histology: lymphocytic infiltrate on labial salivary gland biopsy 6—Postive autoantibodies (anti-Ro/SS-A or anti-La/SS-B Ab, antinuclear Ab, rheumatoid factor) |
| Primary Sjögren’s syndrome: definite: 4 criteria; probable: 3 criteria Secondary Sjögren’s syndrome: 4 criteria (excluding item 6) |
Table 9.4 Autoimmune diseases associated with secondary Sjögren’s syndrome
| Connective tissue disorders: rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), progressive systemic sclerosis, polymyositis, polyarteritis nodosa, mixed connective tissue disease, MacDuffie’s syndrome. |
| Other rheumatic disorders: psoriasis, Behçet’s disease, giant-cell arteritis. |
| Autoimmune diseases: Hashimoto thyroiditis, primary biliary cirrhosis, chronic active hepatitis, hypothyroidism and hyperthyroidism. |
| Miscellaneous: thrombocytopenic purpura, Biermer’s disease, Crohn’s disease, hypergammaglobulinemia, Waldenström’s macroglobulinemia, celiac disease, myasthenia, Sweet’s syndrome, lipodystrophy. |
There is no real consensus on the definition of Sjögren’s syndrome, which is increasingly considered as a clinical entity situated at the crossroads of systemic diseases, malignant diseases (especially hematological), and viral diseases. There is a strong gender predilection in Sjögren’s syndrome, as 90% of middle-aged patients are women. Initially defined as the xerophthalmia-xerostomia-rheumatoid arthritis triad, its definition changed when it was realized that other systemic diseases could be associated.18 Primary Sjögren’s syndrome is now defined as the isolated syndrome, with xerophthalmia-xerostomia in the biological and immunopathological context described below.19 However, multiorgan involvement is often found, in the form of connective tissue disorders. The dividing line between primary and secondary Sjögren’s syndrome is often hazy. Secondary Sjögren’s syndrome is defined as a combination of signs of Sjögren’s syndrome and a systemic disease (Table 9.3).20 Although xerophthalmia and xerostomia are the main symptoms, multiorgan involvement very frequently provides the presenting signs of the disease. HLA-DR3, -DRW52, -DRW3 is associated with primary Sjögren’s syndrome, and HLA-DR4, -DRW3 with secondary Sjögren’s syndrome. All connective tissue diseases can be accompanied by a sicca syndrome: lacrimal and/or salivary involvement, although nonspecific, is part of the pathology of connective tissue diseases21(Table 9.4).
Primary Sjögren’s syndrome is often more severe than secondary Sjögren’s syndrome. The main difficulty is in distinguishing, among patients with tear insufficiency (16% of the population over 80 years), those with an authentic Sjögren’s syndrome likely to benefit from appropriate therapy. Sjögren’s syndrome, be it primary or secondary, is one of the causes of the sicca syndrome associated with lymphocyte infiltration. The infiltrating cells in Sjögren’s syndrome differ from those in other sicca syndromes, which include sarcoidosis, graft-versus-host disease, human immunodeficiency virus (HIV) infection, hemochromatosis, and amylosis, and must therefore be considered as Sjögren-like conditions.
The systemic manifestations of Sjögren’s syndrome are linked to direct lymphocyte toxicity for various tissues. They include thyroiditis, hepatitis, primary biliary cirrhosis, celiac disease, and various pulmonary, cutaneous, or nervous system diseases. Anti-Ro (SS-A) and anti-La (SS-B) antibodies are frequently present. However, these antibodies are not at all specific, as they also exist in other diseases such as SLE. Their titer can vary independently of clinical severity.
Ocular involvement is present in approximately 90% of patients with Sjögren’s syndrome, but its expression is variable. Relatively mild at the outset, the symptoms include a foreign-body sensation, a burning sensation, photophobia, and redness. Patients sometimes also complain of difficulty in opening their eyes in the morning, and this can become permanent in severe forms with complications such as filamentary keratitis, corneal ulcerations, and symblepharon formation. Infections are mainly linked to a failure of lacrimal defense mechanisms.
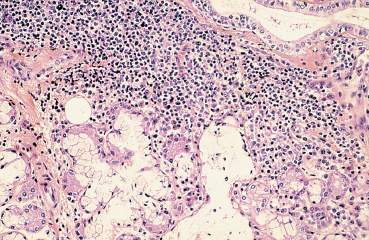
Clinical examination shows an abnormal tear film: the tear meniscus is reduced and there are abundant mucous debris and strands of rubbery mucus on the conjunctiva and cornea. The Schirmer I test is subnormal (less than 5 mm in 5 mins) (Fig. A) and the tear break-up time is short (below 10 s) (Fig. B). Punctate corneal epithelial fluorescein staining is often most intense in the inferior cornea and in the interpalpebral exposure zone (Fig. C). Contrary to common belief, rose bengal does not directly stain dead cells but rather reflects the quality of the tear film by revealing areas of the ocular surface not covered by mucus.22,23 Rose bengal and lissamine green (less painful) are more specific and sensitive (Figs. D and E),24 but the results are meaningless unless the score is above 3.5 in the Van Bijsterveld grading scale. It can be useful to study tear osmolarity, which is significantly increased in patients with tear deficiency, but an experienced technician is required. Conjunctival impression cytology shows a reduction in goblet cells and squamous metaplasia associated with a change in the nuclear/cytoplasmic ratio.25,26 However, classical snake-like chromatin in conjunctival cells is not at all pathognomonic for sicca syndrome. Low lysozyme and lactoferrin levels are a supplementary diagnostic argument in favor of Sjögren’s syndrome. Parotid sialography is less contributory than scintigraphy of the salivary glands. Biopsy of accessory salivary glands remains the most sensitive and most specific examination, even if the number of false-negative results remains high (Fig. F).
Currently, there is no consensus on the diagnostic criteria of Sjögren’s syndrome. Diagnosis is therefore based on a combination of clinical and laboratory findings.
Diagnostic laboratory tests: antinuclear Ab, rheumatoid factor, SS-A and SS-B Ab. Biopsy of accessory salivary glands.
Vasculitides
Wegener’s granulomatosis is a necrotizing granulomatous vasculitic disease involving, in decreasing order of frequency, the lungs (95%), sinuses and nasopharynx (90%), and kidneys (85%). Ocular involvement occurs in an estimated 28-45% of cases.
The basic histopathological lesion is granulomatous necrosis, frequently associated with vasculitis identical to that seen in polyarteritis nodosa.27,28
Involvement of the eye and its adnexa can reveal the disease. Peripheral ulcerative keratitis, scleritis (especially nodular or necrotizing [Fig. 9.7]), and conjunctival inflammation (4-17%) are all early signs. The diagnosis can be confirmed by biopsy of ocular tissue to detect the characteristic histopathological lesions (Fig. 9.8).29,29a Detection of serum antibodies to polymor-phonuclear cell cytoplasm (antineutrophil cytoplasmic antibodies [ANCA]) is an excellent diagnostic test, being both sensitive and specific. These antibodies are present in 90% of patients in the active phase and 75% of patients with a limited form of the disease. Their titer, which correlates with the severity of the disease, also appears to be a fairly reliable prognostic indicator.
 Ocular Surface in Dry Eye Syndrome
Ocular Surface in Dry Eye Syndrome
Calculation of a “tear function index,” based on the dilution time of a known amount of fluorescein, is an interesting approach to measuring tear secretion.1,2,3 It seems that tear flow is normal (about 0.10μL/min) in patients with dry eye syndrome. In contrast, assessment of tear evaporation in these patients is controversial.
Several modifications of the conjunctival epithelial surface are induced by dry eye syndrome.
—Goblet cells are sensitive to local electrolyte concentrations. Dry eye syndrome appears to facilitate tear hyper-osmolarity through a rise in the sodium concentration, and would thereby contribute to a reduction in goblet cell density1 or their loss. At the beginning there is a transient rise in the density of goblet cells. In contrast, the “second mucus system” is not altered.2
—Mucin acid glycoproteins become neutral. This leads to a reduction in the expression of certain oligosaccharides such as sialic acid, N-acetyl-chondrosamine, N-glucosamine, and galactose-acetyl-chondrosamine, while mannose levels are increased.3
—The morphology of epithelial cells is relatively unmodified, except for the loss of microvilli.4 The epithelial cells increase in size and become polygonal, with a reduction in the nuclear-cytoplasmic ratio. The nuclear chromatin condenses, taking on a “snake-like” appearance.5 Conjunctival squamous metaplasia and decreased goblet cell density are not, however, directly caused by the reduction of tear secretion.6
—Corneal sensitivity decreases, partly due to histopathological changes induced by tear insufficiency.3
—Inflammatory mediators contribute to the ocular surface modifications due to tear insufficiency. Their presence leads to a rise in the corresponding markers (HLA-DR, ICAM-1, and IL-6). The responsibility of Epstein-Barr virus has not been confirmed in recent studies.7,8
References
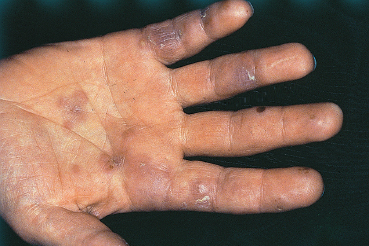
Fig. 9.9 Polyarteritis nodosa: vasculitic lesions on the fingers and palm
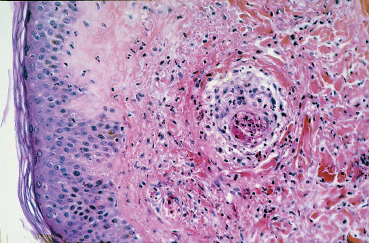
Fig. 9.10 Polyarteritis nodosa: perivasculitis associated with fibrinoid necrosis of the arteriole wall in the dermis (light microscopy, HES x 400) (Courtesy of Professor B. Kantelip, Besançon)
Diagnostic laboratory test: ANCA.
Polyarteritis Nodosa
Polyarteritis nodosa is a rare disease that mainly affects the small and medium-sized arteries (Figs. 9.9 and 9.10). The clinical picture depends on the affected vessels and their distribution. Myalgia, polyneuritis, and nephropathy are the most frequent complications.30 Twenty per cent of patients with polyarteritis nodosa have ocular involvement, mainly of the retina. Orbital inflammation in the form of exophthalmia and, more frequently, peripheral ulcerative keratitis, are other ocular manifestations. Conjunctival infarction characterized by localized areas with a waxy hue, and true necrosis with subcon-junctival hemorrhage and chemosis, are rarer.31
Diagnostic laboratory tests: blood tests of inflammation, hepatitis B antigen (50%), electromyogram, and muscle biopsy.
Giant Cell Arteritis
Giant cell arteritis is a form of subacute inflammatory panar-teritis of the elderly predominating in the region of the brain. Headache is the chief symptom. Ocular signs, mainly of ischemic origin, affect one in five patients.32,32a Conjunctivitis can be present, especially in patients with (rare) inflammatory pseudotumors. In such cases it is associated with ptosis, lid edema, and chemosis. Scleritis, episcleritis, and peripheral ulcerative keratitis are other potential ocular complications.
Diagnostic laboratory tests: erythrocyte sedimentation rate, C-reactive protein, biopsy of the temporal artery.
Metabolic Disorders and Intoxications
Amylosis
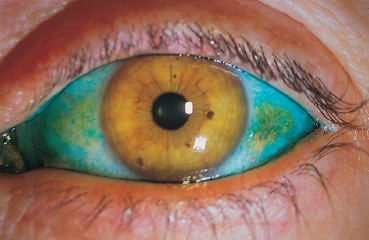
Amylosis is a disease characterized by deposits of an insoluble protein in various organs. In generalized amylosis, ocular involvement can be isolated or associated with systemic involvement.33 Nodular yellowish conjunctival infiltrates with hemorrhage and keratoconjunctivitis sicca due to infiltration of the lacrimal gland are possible, but corneal amylosis is more frequent. In localized conjunctival amylosis the conjunctiva appears waxy and poorly vascularized, and yellow-orange nodules can be found in the upper fornix.
Diagnostic laboratory tests: conjunctival biopsy, rectal biopsy.
Vitamin Deficiencies
Vitamin A being crucial for the growth and differentiation of epithelial cells, its deficiency is responsible for many cases of blindness worldwide. Conjunctival involvement is isolated in the initial stages of vitamin A deficiency, with onset of classical xerosis (Bitot spots) linked to a gradual loss of goblet cells. Later stages are complicated by corneal involvement.34
Deficiencies in other vitamins (B1, B6, etc.) can also cause conjunctival disorders, such as moderate keratoconjunctivitis sicca. The ocular dryness observed during anorexia nervosa is probably due to vitamin deficiency.
Diagnostic laboratory tests: plasma retinol assay (rarely available in developing countries, which are most affected).
Endocrine Diseases
Hyperthyroidism
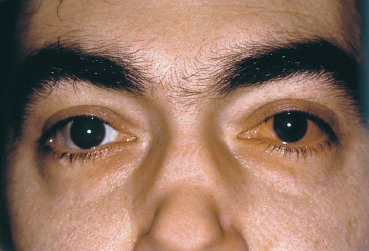
Ocular signs frequently reveal thyroid dysfunction. The main form of thyroid dysfunction is Graves disease, defined by the triad of hyperthyroidism, ophthalmopathy, and cutaneous involvement. Conjunctival manifestations may be limited to sectorial or diffuse hyperemia at the level of the horizontal muscles, but chemosis preventing lid closure can also occur.35 Graves’ ophthalmopathy can be associated with superior limbic keratoconjunctivitis (see Chapter 10).36 Exophthalmia is mainly linked to the autoimmune disorder, which is responsible for muscle inflammation, edema, infiltration of orbital fat, and reactive fibrosis. Chemosis can be due to venous congestion induced by the rise in the volume of intraorbital structures.
Hypoparathyroidism
Linked to parathyroid hormone deficiency, hypoparathyroidism is associated with systemic signs of hypocalcemia. The most characteristic form of ocular involvement is tetanic sub-cortical cataract. However, some patients can also develop keratoconjunctivitis sicca.
Multiple Endocrine Neoplasms
Multiple endocrine neoplasms are malignant tumors that generally appear during the third decade. Ocular involvement is characterized by neuromatosis of the bulbar and palpebral conjunctiva. These neuromas are frequent (87%), and take the form of enlarged nerves that are arranged in bundles and are especially visible at the corneoscleral limbus. Involvement of the palpebral conjunctiva gives the lid a thickened appearance (plexiform neuroma), with smooth yellowish tumors visible at the lid margin in the form of bundles of axons. Ocular dryness is also frequent (67 %). It is important to detect these signs, not only because it permits early treatment of these diseases (sometimes before malignant transformation), but also because it permits family screening.
Diseases of the Digestive Apparatus
Hepatic Diseases
Hepatic diseases are numerous but their ocular complications are rare. The latter are mainly linked to a serum deficiency in vitamin A. Poor adaptation to darkness and xerophthalmia are thus the most frequent manifestations.37
Primary Biliary Cirrhosis
Primary biliary cirrhosis is a rare autoimmune disease consisting of chronic nonsuppurative necrotizing cholangitis. The clinical signs are those of cholestasis (jaundice and pruritus), and Raynaud’s phenomenon is frequent. Sjögren’s syndrome is present in 36-73% of patients with primary biliary sclerosis, according to a study.38
Diagnostic laboratory tests: antimitochondrial Ab (present in 95 % of cases when primary biliary cirrhosis is associated with Sjögren’s syndrome).
Hemochromatosis
This hereditary autosomal recessive disease with variable penetrance is accompanied by iron malabsorption. Thesauris-mosis occurs on the conjunctiva (which is covered with pseu-dopigmentary deposits) and on the lacrimal gland, leading to a dry eye syndrome.
Diagnostic laboratory tests: hypersideremia, rise in trans-ferrin saturation, needle biopsy of the liver.
Hepatitis
A dry eye syndrome is especially frequent in patients with hepatitis involving an autoimmune mechanism, but an authentic dry eye syndrome has also been found during active hepatitis C.39
Diagnostic laboratory test: anti-HCV Ab.
Gastrointestinal Diseases
Crohn’s Disease
Crohn’s disease affects the entire intestine but particularly the colon and ileum. While gastrointestinal disorders usually reveal the disease, it can also be diagnosed on the basis of extra-gastrointestinal signs. Ocular involvement during Crohn’s disease is far from rare, affecting 10-30% of patients. Granulomatous episcleritis, which is directly linked to the disease, is a sign of progression, while xerophthalmia is secondary to vitamin A malabsorption.40
Diagnostic laboratory test: coloscopy, intestine biopsy.
Whipple’s Disease
Whipple’s disease is a rare disease of infectious origin with duodenal and central nervous system complications. Ocular involvement, with chemosis, tearing, and filamentary keratitis sometimes reveal the disease.41
Diagnostic laboratory test: polymerase chain reaction (PCR).
Blood Diseases
Graft-Versus-Host Disease
Graft-versus-host disease occurs after allogenic bone marrow transplantation, probably because of mismatched minor histocompatibility antigens. It is due to a reaction of the graft against its immunocompetent host, and occurs in acute and chronic forms.
The acute form occurs approximately 20 days after the transplant. The three target organs are the skin, the liver, and the digestive tract. Conjunctivitis is sometimes present, consisting of simple hyperemia or acute pseudomembranous conjunctivitis with authentic sloughing of the corneal epithelium. The intensity of the conjunctival involvement appears to be a good prognostic index of the rejection process.42
During chronic rejection, approximately 11 % of patients develop conjunctival involvement which resembles the fibrosing conjunctivitis of ocular cicatricial pemphigoid. The pathognomonic sign of passage to chronicity is the formation of a fibrous line on the tarsal conjunctiva.43 The lacrimal glands are very frequently involved, leading to a generally irreversible sicca syndrome in about 50% of patients.
Conjunctival Complications of Immunosuppressive Chemotherapy
Toxic conjunctivitis induced by cytotoxic immunosuppressive drugs (Table 9.5) should be distinguished from infectious conjunctivitis. It is generally acute, but repeated treatment can hinder the diagnosis.44
Infectious Diseases
Brucellosis
Brucellosis is due to a bacterium belonging to the gender Bru-cella. The main reservoir is animals, and especially cattle. Ocular involvement consists of phlyctenular conjunctivitis and nummular subepithelial keratitis.46
Diagnostic laboratory test: serology.
Table 9.5 Conjunctival complications of cytotoxic immunosuppressive chemotherapy45
| Cyclophosphamide | Blepharoconjunctivitis |
| 5-fluoro-uracil | Canaliculus infiltration and fibrosis |
| Cytosine-arabinoside | Conjunctivitis |
| Methotrexate | Conjunctivitis |
| Doxorubicin | Conjunctivitis |
| Deoxycoformycin | Conjunctivitis |
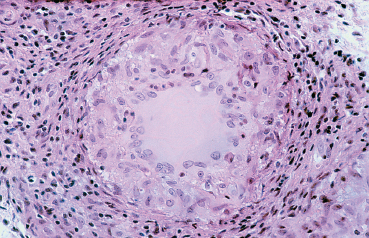
Fig. 9.11 Tuberculosis: granuloma composed of epithelioid cells and Langhans giant cells (light microscopy, HESx400) (Courtesy of Professor B. Kantelip, Besançon)
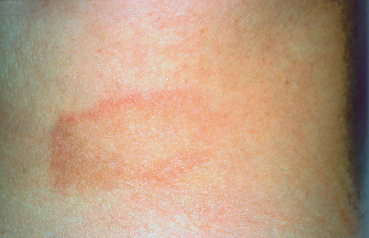
Fig. 9.12 Lyme disease: “erythema chronicum migrans” (Courtesy of Dr. S. Dalac, Dijon)
Table 9.6 Causes of Parinaud’s oculoglandular syndrome47
| Cat-scratch disease, sporotrichosis, tuberculosis, syphilis, tular-emia, coccidioidomycosis, actinomycosis, blastomycosis, pasteur-ellosis, yersiniosis, listeriosis, mumps, lymphogranuloma venereum, sarcoidosis, Newcastle disease |
Tularemia
Tularemia is a zoonosis due to Francisella tularensis. The main vector is the rabbit, although transmission can also occur via other routes such as insect bites. The chief symptoms are fever, nausea, vomiting, and headache. In adults and children the ocular involvement is characterized by Parinaud’s oculoglandular syndrome. The conjunctivitis is unilateral and composed of multiple yellow tarsal conjunctival granulomas, that are large, sometimes ulcerated, and associated with preauricular, submandibular, or cervical lymphadenopathies (Table 9.6).
Diagnostic laboratory test: agglutination tests, detection of the pathogen in lymph node serosities.
Tuberculosis
Tuberculosis is due to Mycobacterium tuberculosis. It mainly affects the lungs, but the entire body can be involved (Fig. 9.11). There were hopes that tuberculosis could be eradicated, but the increasing number of immunocompromised patients has led to the emergence of atypical forms due to other my-cobacterial strains. The incidence of ocular involvement is about 1-2%. It can be directly linked to infection by the pathogen, but can also be secondary to a hypersensitivity reaction to circulating tuberculin protein. The conjunctival manifestations mainly include redness, tearing, and mucopurulent secretions. Unilateral ulcerated or nodular conjunctivitis can also occur.48 Phlyctenular keratoconjunctivitis appears to be due to a hypersensitivity reaction. In older patients the conjunctivitis can be bilateral and is often associated with lymphadenopathy. Tuberculosis is one cause of Parinaud’s oculoglandular syndrome (Table 9.6).
Diagnostic laboratory test: skin test, chest radiograph, histological examination, and culture of tissues.
Leptospirosis
Leptospirosis is due to a spirochete and is mainly transmitted by rats. Characterized by a two-phase course, it first causes a febrile malaise of abrupt onset associated with muscle pain and a morbilliform eruption, followed by signs so varied that they often mislead the diagnosis. Conjunctival involvement occurs during the first phase and takes on a fairly characteristic form of conjunctival effusion which is often ignored. Engorged conjunctival vessels and anastomoses with episcleral vessels typically occur.
Diagnostic laboratory tests: blood culture in the early phase, then serology.
Lyme Disease
Due to Borrelia burgdorferi spirochetes, this highly proteiform disease is transmitted by ticks. It is characterized by a skin rash (Fig. 9.12) which is then complicated by general malaise and sometimes by articular, neuromeningeal, and cardiac manifestations. All the ocular components can be affected, uveitis being the most frequent manifestation. Conjunctivitis, present in 11 % of cases, generally occurs shortly after the skin rash but is generally mild.49 Some patients have chronic follicular forms. It is not known whether the ocular involvement is linked directly to the spirochete or to a hypersensitivity reaction. Diagnostic laboratory test: serology, Western immunoblot.
Syphilis
Syphilis is due to Treponema pallidum. The advent of penicillin led to a fall in the frequency of this disease, which was a source of many misdiagnoses. However, its frequency has been rising again in the last decade and syphilis takes on a particularly deceitful clinical aspect in immunocompromised patients. All the ocular tissues can be affected during the three main phases of the disease. In the primary stage the conjunctiva can be affected in the form of papillary conjunctivitis or a painless ulcerated chancre with indurated edges. The latter is typically unilateral, located on the lower bulbar conjunctiva, associated with locoregional lymphadenopathies, and leaves a scar. During the secondary phase, mucous patches with a phlyctenular aspect, gray-white nodules, papillary conjunctivitis, or tarsitis can occur. The tertiary phase is mainly associated with granulomatous conjunctivitis, tarsitis, and gummae. In congenital syphilis the ocular signs are the same as during the secondary and tertiary phases.
Diagnostic laboratory test: serology.
Acquired Immunodeficiency Syndrome (AIDS)
Ocular involvement, clinically present in 75% of cases, can be linked to four mechanisms: vascular disorders, opportunistic infections, malignancies, and neurological abnormalities. Vasculitis preferentially affects the retinal vessels but the conjunctiva can also be the site of microangiopathy. The conjunctiva can harbor lesions of Kaposi’s sarcoma, a multifocal vascular tumor that mainly affects the conjunctiva, the lid margins, and more rarely the socket. Eighteen per cent of patients with AIDS who have Kaposi’s sarcoma also have ocular or periocular involvement.51 The conjunctival lesions are situated in the lower fornix and develop slowly in the form a bright red mass sometimes mimicking subconjunctival hemorrhage.
Blepharoconjunctivitis can occur if the palpebral lesions become ulcerated and superinfected.52 Microsporidiosis is another cause of chronic keratoconjunctivitis in patients with AIDS. It is a ubiquitous obligate, intracellular, spore-forming protozoan. Conjunctival involvement consists of bilateral follicular-papillary conjunctivitis, and is associated with a characteristic diffuse “macropunctate” epithelial keratopathy which is slowly progressive. In immunocompetent patients, the inflammation is more severe and associated with stromal keratitis. The diagnosis is confirmed by electron microscopic examination of biopsy specimens which reveals the organisms.52a,52b Other frequent ocular disorders are abnormalities of the conjunctival vessels, with telangiectasias, abnormally tortuous and dilated vessels, and hemorrhage. A sicca syndrome occurs in 15% of cases.
Pharyngeal Conjunctival Fever (see Chapter 6)
Pharyngeal conjunctival fever is a highly contagious acute disease progressing in epidemic form and mainly due to type 3 adenovirus. It is characterized by symptoms similar to those of epidemic keratoconjunctivitis but differs from the latter by the presence of systemic signs (Table 9.7).
Conjunctival signs are varied, ranging from simple itchiness to a severe burning sensation associated with inflammation and tearing. Photophobia is generally mild. Characteristically, follicles predominate on the palpebral conjunctiva, where lymphoid tissue is most abundant. They are accompanied by petechial hemorrhages that can sometimes cause impressive subconjunctival hemorrhaging. Pseudomembranes can occur, although they are rarer than in epidemic keratoconjunctivitis. Pseudomembranes are mainly indicative of adenovirus or streptococcal infection, which is more frequent in children. Corneal involvement is not always present, and consists of sub-epithelial infiltrates.
Immunological Diseases
Polymyositis-Dermatomyositis
Polymyositis causes proximal muscle fatigability. It can be isolated or associated with another systemic disease or malignancy.53 The typical heliotropic blanching of the upper eyelids, a facial erythematous butterfly rash and periorbital edema are the most frequent signs. Conjunctival edema is present in 70% of cases. Membranous conjunctivitis and true occlusion of conjunctival vessels are also seen.
Dermatomyositis is a form of polymyositis associated with cutaneous involvement.
Diagnostic laboratory tests: rise in muscle enzyme levels, electromyogram (myogenic pattern), muscle biopsy (focal inflammatory infiltrate, areas of necrosis replaced by regenerated muscle tissue).
Table 9.7 Comparison of pharyngeal conjunctival fever (PCF) and epidemic keratoconjunctivitis (EKC)
| Parameter | PCF | EKC |
|---|---|---|
| Age | <10 years | 20-40 years |
| Incubation | 3-12 days | 3-12 days |
| Serotypes | 3,4,7 | 8,19 |
| Follicular conjunctivitis | +++ | +++ |
| Preauricular adenopathy | + | ++ |
| Keratitis | +/- | +++ |
| Pharyngitis | +++ | – |
| Fever | +++ | – |
| Pseudomembrane | +/- | + |
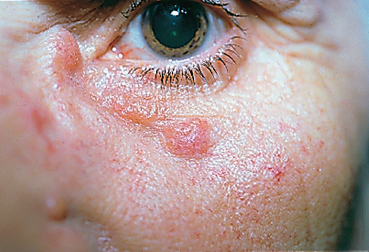
Fig. 9.13 Sarcoidosis: palpebral sarcoids
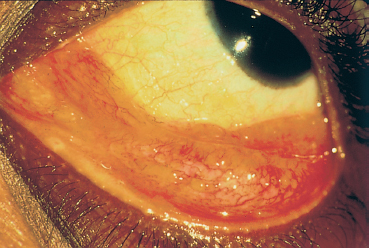
Fig. 9.14 Sarcoidosis: sarcoids of the conjunctival fornix
Sarcoidosis
Sarcoidosis is an idiopathic granulomatous disease mainly affecting young adults and involving many organs. It is associated with lymphadenopathies, mainly within the thoracic cavity. Sometimes asymptomatic (36% of cases), it leads to ocular signs in 19% of cases at initial presentation54 (Fig. 9.13). Anterior uveitis is by far the most frequent manifestation. The conjunctiva can present low, gray-yellowish nodules mainly located in the inferior fornix (Fig. 9.14) or the posterior surface of the lower lid, in approximately one third of patients. Their biopsy shows noncaseating granulomatous lesions in 30% of cases (Fig. 9.15).55 Keratoconjunctivitis sicca is linked to granulomatous infiltration of the lacrimal glands. Mikulicz syndrome is rarer, associating hypertrophy of the lacrimal gland and salivary glands.
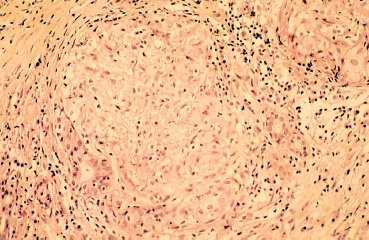
Fig. 9.15 Sarcoidosis: lacrimal gland noncaseating granulomatous infiltration consisting of lymphocytes and epithelioid and giant cells. Only few excretory ducts are spared (light microscopy, HES x 400) (Courtesy of Professor B. Kantelip, Besançon)
Diagnostic laboratory test: angiotensin-converting enzyme (ACE), serum lysozyme, gallium scan, chest radiograph, chest CT-scan and MRI, skin tests, pulmonary function tests, bronchoalveolar lavage, tissue biopsies.
Cogan’s Syndrome
Cogan’s syndrome is characterized by concomitant involvement of the eye and inner ear resembling Ménière’s disease. Other associations with systemic diseases such as Crohn’s disease and sarcoidosis have been reported.56 The origin of this disease is unknown but an infectious cause is suspected. Patients mainly complain of red eyes, discomfort, and photophobia. Decreased vision, tearing, diplopia, or a foreign-body sensation can also occur. Ocular involvement consists mainly of interstitial keratitis (72%) but also of conjunctivitis and rare conjunctival nodules.57
Skin Diseases: Psoriasis
Bullous diseases are not dealt with here, as they are specifically covered in Chapter 4.
Psoriasis is a very frequent cutaneous disease (an estimated 2% of Americans are affected). Cutaneous manifestations consist of red patches covered by scales on the scalp and skin overlying the joints. Joint involvement is sometimes present, associated with the HLA-B27 antigen. Ocular involvement, seen in 10% of cases, can take several forms58: red patches on the eyelids and conjunctiva, blepharitis, and conjunctival irritation ranging from simple redness to true scars (symblepharons). The conjunctivitis can take on a major catarrhal aspect, especially during exacerbations of arthritis. Granulomatous lesions of the eyelids and bulbar conjunctiva can occur. A dry eye syndrome is found in 19% of patients with psoriasis.
Differential Diagnosis
This section focuses on chronic diseases of the conjunctiva that can lead to a false diagnosis of conjunctivitis; they comprise abnormal coloration of the conjunctiva, and some tumors.
Gout
Gout is responsible for renal and articular manifestations due to deposits of uric acid crystals. Ocular involvement mainly comprises uveitis and a rise in intraocular pressure. However, persistent and bilateral dark red coloration of the bulbar and palpebral conjunctiva is present. The origin of this redness is not known.
Porphyria
Porphyrias are diseases linked to an enzyme deficiency that leads to abnormal hemoglobin synthesis. Ocular involvement is infrequent and limited to the eyelids and other exposed areas. In addition to exposure keratoconjunctivitis secondary to a retractile ectropion, conjunctival vesicles can cause severe conjunctivitis with symblepharon formation and cicatricial entropions (as in cicatricial pemphigoid).
Hematological Disorders
Ocular involvement in this setting is mainly linked to deficiencies in clotting factors or blood cells. The manifestations are mainly retinal; conjunctival complications are rare in patients with hematological disorders. Subconjunctival hemorrhage is possible during severe thrombocytopenia or clotting disorders. In dysproteinemias such as myeloma and Waldenström disease the conjunctiva can bear signs of blood hyperviscosity or crystals corresponding to deposits of immunoglobulin and kappa light chains. The emergence of these crystals is not necessarily a sign of malignancy, as benign monoclonal gam-mopathies (that may be precursors of malignant gam-mopathies) are accompanied by similar deposits of para-proteins or cholesterol.
Polycythemia
Polycythemia, be it primary or secondary, corresponds to a rise in blood cells and blood viscosity, leading to a reduction in blood flow. The poorly specific nature of the manifestations often leads to a false diagnosis of neurasthenia. The intense red color of the skin is particularly marked, however. The vivid red color of the conjunctiva generally follows that of the skin.
Malignant NonHodgkin’s Lymphoma
Some lymphomas (conjunctival-associated lymphoid tissue [CALT]-lymphomas) have an isolated conjunctival location (Fig. 9.16). This generally salmon-pink, crescent-shaped tumor can perfectly mimic chronic conjunctivitis at the outset. Its characteristic appearance and persistence should make the practitioner suspicious of the diagnosis. The tumor must be carefully resected, and appropriately processed for immuno-logical characterization of the lymphomatous disease (Fig. 9.17).
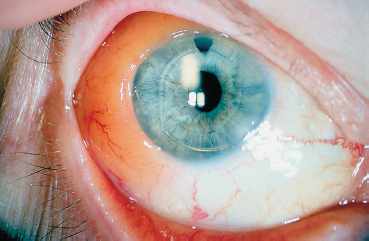
Fig. 9.16 Conjunctival-associated lymphoid phoma: salmon-pink conjunctival tumor
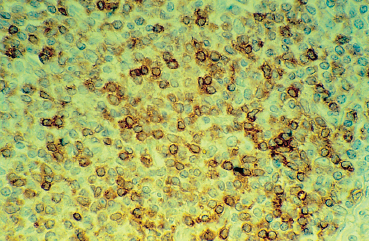
Fig. 9.17 CALT-lymphoma: monoclonal lymphocytic proliferation shown by perinuclear staining with anti-kappa light chain antibodies (light microscopy, frozen section, x 400)
Immune Deficiencies other than AIDS
Patients with B cell deficiencies lack antibodies and therefore have an increased risk of conjunctival infection, especially by Haemophilus influenzae and Staphylococcus epidermidis. 59 Chronic mucocutaneous candidiasis is linked to a specific deficiency in lymphocytes targeting Candida species. Infectious blepharitis and chronic keratoconjunctivitis can result.
Kawasaki Disease
Kawasaki disease is a childhood disease characterized by an exanthematous eruption and multisystem involvement. The diagnosis is based on the presence of five of the following six criteria: fever, bilateral conjunctival injection, modification of the respiratory mucosa, modifications of the nails and the skin of the extremities, maculopapular eruption, and cervical lym-phadenopathy. More than 90% of children with Kawasaki disease have nonexudative vasodilation of the conjunctival vessels, which is bilateral and mainly affects the bulbar conjunctiva60 (Fig. 9.18). There is no chemosis, follicles, papillae, or preauricular node enlargement. The conjunctival involvement usually resolves without sequelae within 2-4 weeks. Kawasaki disease has been designated “childhood polyarteritis nodosa.” The diagnosis is mainly clinical.
Kidney Disorders
Kidney disorders are accompanied by the manifestations of the causal disease. This section focuses on disorders of phos-phocalcium metabolism that lead to calcium phosphate and apatite crystal deposition in the cornea and conjunctiva in the interpalpebral exposure zone, where alkalinization due to hypercapnia may play a role by reducing the solubility of calcium phosphate. However, it is not certain that the ocular redness observed during renal failure is directly linked to the presence of these crystals.61
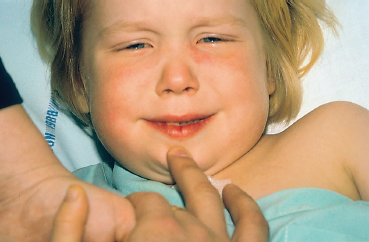
Fig. 9.18 Kawasaki disease: erythematous edema of the face, cheilitis, erythema of the cheeks, and conjunctivitis (Courtesy of Dr. S. Dalac, Dijon)
Hereditary Hemorrhagic Telangiectasia: (Rendu-Osler-Weber Syndrome)
Rendu-Osler-Weber syndrome is a hereditary condition characterized by the onset of multiple telangiectases of the skin and mucosa, and visceral arteriovenous malformations, particularly in the pulmonary circulation. Ocular involvement is rare and consists of conjunctival telangiectasias and pete-chiae, angiomatous nodules, or cobweb formations, which can cause subconjunctival hemorrhage.62
Practical Management
Systemic disease is rarely diagnosed on the basis of conjunctival signs alone, and the different conjunctival lesions have no pathognomonic significance. More interestingly, a combination of a sometimes mild conjunctival disorder with involvement of another organ can lead to the diagnosis and therefore to appropriate systemic therapy. There follows a list of certain clinical presentations that should arouse suspicion and sometimes call for complementary investigations.
Conjunctivitis and Ear, Nose, and Throat-Involvement (Deafness, Sinusitis)
Deafness-dizziness: Cogan’s syndrome
Sinusitis: Wegener’s granulomatosis
Chondritis-deafness-dizziness: relapsing polychondritis
Conjunctivitis and Fever
Infectious diseases (rickettsiosis, leptospirosis, brucellosis, etc.), ear, nose, and throat (ENT) infections
All systemic diseases in the acute inflammatory phase
Conjunctivitis and Arthralgia
All causes of xerophthalmia
Reiter’s syndrome
Brucellosis
Polyarteritis nodosa
Wegener’s granulomatosis
Relapsing polychondritis
Dry Eye Syndrome and Arthralgia
Sjögren’s Syndrome
—Primary (without an associated autoimmune disease)
—Secondary (associated with RA, SLE, progressive systemic sclerosis, autoimmune hemolytic anemia, Hashimoto thyroiditis, etc.), with or without lymphoma
Dacryoadenitis and Parotiditis
Hodgkin’s disease
Lymphoma
Sarcoidosis
Tuberculosis
Amylosis
Crohn’s disease, hepatitis C, graft-versus-host disease
Recurrent Infectious Conjunctivitis (No Local Cause)
Acquired hypogammaglobulinemia (nephrotic syndrome, CLL, Sjögren’s syndrome)
Complement deficiency
Conjunctivitis and Gastrointestinal Disorders
Crohn’s disease
Whipple’s disease
Ulcerative colitis
Polyarteritis nodosa
Infectious diseases
Conjunctivitis and Raynaud’s Phenomenon
Progressive systemic sclerosis
Systemic lupus erythematosus
SHARP syndrome
Primary biliary cirrhosis
Conjunctivitis and Skin Conditions
Kawasaki disease
Psoriasis
Systemic lupus erythematosus
Autoimmune mucocutaneous bullous diseases
Rosacea
Allergy
Granulomatous Conjunctivitis and Lymphadenopathies
Tularemia
Cat-scratch disease
Tuberculosis
Syphilis
Coccidioidomycosis
Pasteurellosis, yersiniosis, listeriosis
Mumps
Sarcoidosis
Pharyngeal conjunctival fever
Indications of Conjunctival Biopsy (see Chapter 2, Focus 4, Chapter 4, and Chapter 8)
It can provide the diagnosis in some circumstances when it reveals specific histopathological lesions, for example, in progressive systemic sclerosis, sarcoidosis, tuberculosis, Wegener’s disease, infections, and autoimmune conjunctivitis.
It offers the opportunity to study the conjunctiva throughout its thickness, including the epithelium and chorion. Underlying immunopathological mechanisms can be analyzed by means of direct immunolabeling (immunofluorescence, im-munoperoxidase staining) with light and electron microscopy. Signs of vascular or perivascular disease in patients with chronic conjunctivitis (often associated with scleritis and/or episcleritis) will point to a systemic vascular disease and call for complementary investigations.
It is more difficult to use conjunctival biopsy as a means of following the disease course, because of its invasive nature. Impression cytology then becomes particularly valuable, despite certain limitations on the information it can provide (only superficial cells that are ready to desquamate are sampled).63 Impression cytology is particularly valuable in the Sjögren’s syndrome.64
 Role of Inflammation in Dry Eye Syndrome
Role of Inflammation in Dry Eye Syndrome
Dry eye syndrome is one of the most frequent ocular disorders, but it is generally mild. Approximately 15%ofpeople over 65 suffer from ocular dryness, and 10% regularly use substitutive eyedrops. Dry eye syndrome is often neglected because of its apparent benignity and intractable course, which is frustrating for patients and unrewarding for ophthalmologists. Nevertheless, it is often far from benign, leading to a permanent ocular burning sensation, painful superficial keratitis, decreased vision, and even to progressive corneal opacification and corneal blindness. Therapy is based on repeated and long-term use of topical lubricants but, for want of a causative treatment, efficacy is often only partial. On the contrary, the symptoms may worsen in case of inappropriate, ineffective, or toxic therapy.
In order to treat effectively dry eye syndromes, it is important to identify their pathogenetic mechanisms, especially the role of inflammatory processes, which often is important but underestimated. Inflammation in dry eye syndrome is often infraclinical and hard to detect, its mechanisms are complex and difficult to dissect, and inappropriate therapy may worsen it.
Whatever the initial mechanism, especially hormonal factors, chronic dessication of the ocular surface induces permanent nervous stimulation aimed at triggering regulatory and epithelial healing mechanisms. These neurogenic stimuli provoke the release of inflammatory cytokines into the tear film, the lacrimal glands, and the conjunctiva. Epithelial cells themselves can secrete IL-1-α, IL-6, and IL-8, and tumor necrosis factor alpha (TNF-α)1. Progressive primary or secondary infiltration of the conjunctiva by immune cells provides all the components required for a chronic cytotoxic inflammatory reaction.
These immune reactions of the ocular surface lead to progressive destruction of the lacrimal glands, followed by the conjunctival epithelium (Fig. Ia and Ib) (see Chapter 2, Focus  6), and especially goblet cells.2,3 The postmenopausal loss of androgenic stimulation can facilitate both ocular inflammation and dryness, because of the immunosuppressive properties of androgens involved in tear regulation. Apoptosis of lacrimal gland secretory acinar cells occurs in animals only 4 hours following ovariectomy.4 This apoptosis is very closely linked to immune phenomena, and degenerated cells could be sources of autoantigens and secondary antoim-munization.
6), and especially goblet cells.2,3 The postmenopausal loss of androgenic stimulation can facilitate both ocular inflammation and dryness, because of the immunosuppressive properties of androgens involved in tear regulation. Apoptosis of lacrimal gland secretory acinar cells occurs in animals only 4 hours following ovariectomy.4 This apoptosis is very closely linked to immune phenomena, and degenerated cells could be sources of autoantigens and secondary antoim-munization.
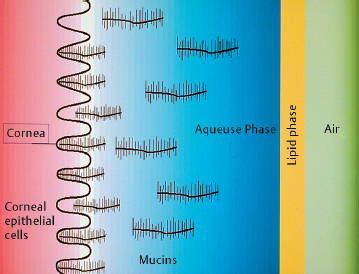
Fig. G Schematic representation of the tear film
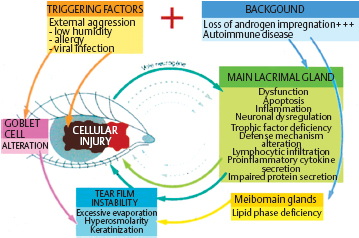
Fig. H Vicious circle of ocular injury in dry eye syndromes
Thus, in both authentic autoimmune conditions and simple postmenopausal lacrimal gland involution, a local inflammatory reaction is present at the ocular surface. Im-munoallergological studies have shown conjunctival inflammation in more than two thirds of patients with dry eye syndrome.5 The inflammation can preexist and induce the dryness, or be associated with it, or occur following allergenic treatments.
Sjögren’s Syndrome (see p. 147 ff)
One of the most characteristic dry eye syndromes of inflammatory origin, although not the most frequent, is that associated with Sjögren’s syndrome. This is an authentic autoimmune disease, affecting not only the main lacrimal gland but also the entire conjunctival surface, which bathes in a tear film rich in inflammatory mediators. Local immune activation leads to conjunctival infiltration by inflammatory cells and directly alters epithelial cell metabolism. This diffuse inflammation of the ocular surface, rather than simple main lacrimal gland involvement, probably accounts for the severity of dry eye associated with Sjögren’s syndrome. Seric auto-antibodies including antinuclear or antiacetylcholine receptor autoantibodies are often detected.6 These latter could block parasympathetic lacrimal gland regulation and thereby inhibit tear secretion.
A viral cause, especially Epstein-Barr virus, has often been put forward but never proved. Similarly, a causal relationship with infection by hepatitis C virus or human T cell leukemia virus type 1, (HTLV-1) (a retrovirus belonging to the same family as the AIDS virus) also may exist. Indeed, HTLV-1 is endemic in tropical countries and is very frequently associated with dry eye or even a true Sjögren’s syndrome.7 Fragments of the viral genome were found in lacrimal gland biopsy specimens, but the responsibility of this virus has not yet been clearly established. Finally, Sjögren’s syndrome can be secondary to systemic autoimmune conditions such as rheumatoid arthritis or collagenoses, and dry eye syndrome can be associated with sarcoidosis or lymphoproliferative syndromes.
Dry Eye Syndrome Associated with Allergy
(see Chapter 3)
Dry eye syndrome is frequently induced or worsened by chronic allergic conjunctivitis, and the two conditions are often interlinked. Allergy is responsible for chronic or recurrent local immune stimulation. The cytokines thus released are cytotoxic for the epithelium and directly alter tear film quality, affecting both the mucin and lipid phases. In particular, they can cause significant loss of goblet cells8, thereby markedly shortening the tear break-up time. Symptoms may be intense, even though conjunctival inflammation is apparently mild.
Typically, marked symptoms often persist for weeks or months after a true allergic episode. Antiallergic treatments are generally ineffective or worsen the symptoms. In such cases it is important to distinguish between the inflammation and secondary ocular dry eye. Many patients can be relieved simply by withdrawing aggressive treatments and by prescribing preservative-free artificial tears and/or gels. This simple therapy breaks the vicious circle due to finger scratching (a mechanical source of mast cell degranulation), dilutes inflammatory mediators, protects the ocular surface from upper lid friction, and helps stabilize the tear film.
Dry Eye Syndrome Associated with Infections
Dry eye syndrome is a frequent complication of viral kerato-conjunctivitis or severe acute or subacute conjunctivitis of other microbial origin. Irreversible goblet cell loss can occur, due to the infectious process and/or its (toxic) treatments, including topical antibiotics, corticosteroids, or cicatrizing agents. Some patients complain that their symptoms persist, and wrongly believe that their condition has not been correctly treated. In fact they have ocular dryness secondary to the infection and often to drug toxicity.
Chronic infectious conjunctivitis is also frequently responsible for severe ocular dry eye, which is all the more difficult to treat when the signs of infection are poorly specific, masked, or ignored. Chlamydial infection is often difficult to diagnose.
Blepharitis and ocular rosacea are also very frequent causes of dry eye syndrome, which includes inflammatory and sometimes infectious components. These disorders are associated with meibomian gland dysfunction, with a resulting change in lipidic phase composition and an increase in tear evaporation. The consequences are a reduced tear breakup time, and epithelial hyperosmolarity. Conjunctival impression cytology shows morphological cell changes, disruption of intercellular junctions, and a loss of goblet cells which probably is responsible for deficient mucin secretion.9 These abnormalities are both a source and a consequence of chronic inflammatory reactions on the ocular surface.
In addition, meibomian dysfunction and poor evacuation of lipid secretions, which are abnormally thick and viscous, facilitate bacterial superinfection at the roots of the lashes. Commensal staphylococci of the lids release lipases, which alter the fatty-acid composition of the meibum and worsen the condition, and toxins, which are often responsible for corneal lesions in the region in contact with the lid margins.
Toxic Dry Eye Syndrome Induced by Eyedrops (see Chapter 8)
Ocular dry eye syndrome induced by degenerative atrophy of the lacrimal glands, although typically mild, may fail to be relieved by artificial tears. Many patients decide to stop using these treatments and feel better as a result. Superficial punctate keratopathy, extending beyond the palpebral fissure or predominating in the inferonasal quadrant, where irritant eyedrops accumulate, points to a toxic action of treatments. Most treatments contain preservatives, which can have direct conjunctival toxicity or occasionally trigger severe local allergy.
All preservatives, to varying degrees, are cytotoxic for the ocualar surface.10,11 Quaternary ammonias also have surfactant properties that modify the lipid phase of the tear film and accelerate its evaporation. They also affect epithelial microvilli, thereby hindering mucus adherence and further destabilizing the tear film. Their cytotoxicity, which is perfectly demonstrated in vitro and in animal models, is reflected by a marked rise in epithelial permeability, which is strongly increased in the course of dry eye syndromes (2.7 times higher than in normal eyes), because of epithelial cell dysfunction. This hyperpermeability can be attenuated by treatment with preservative-free products, but is worsened (by more than 20%) by eyedrops containing benzalkonium chloride, even at low concentrations.12
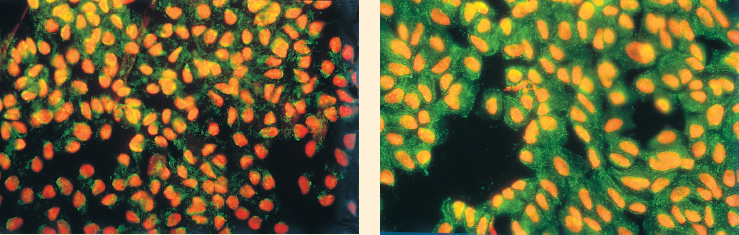
Fig. I Immunolabeling of conjunctival impression cytology, identifying apoptotic cells (APO2.7 marker, in green)
a Weak labeling on a normal eye
b Intense marker expression in a patient with dry eye
In cell culture, very low concentrations of benzalkonium chloride induce apoptotic degeneration, while higher concentrations (as in eyedrops) cause true necrosis, which is compensated for in vivo by epithelial cell turnover. Nevertheless, this chronic cell dysfunction, added to the insult induced by dryness and inflammatory reactions, favors additional immunoinflammatory stimulation. Chronic topical administration of benzalkonium chloride in healthy animals leads to conjunctival inflammatory infiltration and progressive loss of goblet cells; this is also the case in humans after years of topical antiglaucoma treatment.13,14
Therapy
Tailored Therapy
Whether primary, or secondary, inflammatory phenomena play a major role in the pathogenesis of dry eye syndrome. They may exist, even in a white eye where they remain sub-clinical, indicating that sicca syndrome should no longer be considered in all cases as a benign unifactorial disorder. This may explain why topical substitute therapy alone is not sufficient to relieve ocular symptoms of certain dry eye syndromes.
Laboratory investigations which help to identify inflammation are often difficult or poorly specific. Conjunctival scraping, conjunctival impression cytology, IgE detection in tears, or blood tests in search of a systemic inflammatory disease (antinuclear and anti-DNA autoantibodies, SS-A, or SS-B antibodies) can help to guide the choice of treatment.
Blepharitis and rosacea call for meticulous daily lid hygiene to empty the meibomian glands, reequilibrate the lipid component of the tear film, and combat staphylococcal infection. Systemic treatment with tetracyclines can be highly beneficial. Tetracycline therapy in this setting is not strictly “antibiotic”: many staphylococci are resistant, and a short treatment course is ineffective. In contrast, tetracyclines have an anticollagenase action and a regulatory effect on meibomian lipids. They reduce the production of staphylococcal lipases, even by resistant strains, and thereby increase the solubility of meibomian secretions. When combined with lid hygiene, tetracyclines can thus be highly beneficial.
IgE detection in tears points to an allergic condition and thus calls for an allergological work-up, which can lead to eradication of the culprit allergen(s) or to specific desensitization. When the causes are impossible to identify or eliminate, topical antiallergic therapy can be useful. Nevertheless, the presence of preservatives, especially benzalkonium chloride, often limits the efficacy of these products. The patient will benefit most from tear substitutive therapy some time after the allergic episode.
When no cause is found, but clinical or biological signs point to an inflammatory component, an attempt must be made to eliminate preservatives. Prescription of preservative-containing eyedrops is illogical and may be harmful during repeated and prolonged use, particularly in dry eye conditions; unfortunately, however, too few preservative-free solutions are available.
Antiinflammatory and Immunosuppressive Drugs
Corticosteroid therapy has been tested empirically, and sometimes successfully (especially in the Sjögren’s syndrome), but their optimal use and even their real benefit remain to be established. One promising approach is the development of topical cyclosporine preparations; such products are already marketed for veterinary use and very effective in dogs with autoimmune dry eye syndromes. Clinical trials are underway in the United States and Europe in patients with severe sicca syndromes.
Cyclosporine A is an immunosuppressive, noncytotoxic agent initially used in organ transplantation. Its basic action is to inhibit helper T cells (CD4+), while preserving suppressor T cell functions (CD8+). In the eye, topical cyclosporine reaches effective concentrations in the cornea, conjunctiva, and lacrimal gland. It has an antiinflammatory and antiapop-totic action on cells in the lacrimal glands and conjunctival epithelium, while having a proapoptotic action on lymphocytes infiltrating the lacrimal glands, thereby attenuating the inflammatory reaction. Cyclosporine secondarily restores the secretory function of the main and accessory lacrimal glands. It is currently being tested in humans, and may revolutionize the treatment of dry eye syndromes, especially the most severe forms.15
Androgens
Androgens have an immunosuppressive action that is specific for lacrimal glands. They reduce lymphocytic infiltration and inflammation within the lacrimal gland, thereby preventing its degeneration and atrophy. They also stimulate secretion of lacrimal proteins, increase the number of mus-carinic and adrenergic receptors in the lacrimal glands, and reestablish the normal function of meibomian glands.16 Although topical androgens are very effective on sicca syndrome in animals, they are currently being studied in humans because their systemic use induce unacceptable adverse effects such as virilization, especially in women.
The current therapeutic arsenal is large but inconsistently effective. The clinical examination therefore remains fundamental, to determine the type of dry eye and tear-film abnormalities, to search for associated pathogenic mechanisms, to identify therapeutic errors, and to find a way out of the impasse in which both the patient and the physician find themselves. In many cases the discovery of an inflammatory component and the treatment of its underlying cause resolves a difficult situation. In the absence of an causative treatment, it is absolutely essential to withdraw irritant treatments and only maintain tear substitutive therapy with topical lubricants (solutions or gels).
The psychological component must not be overlooked. Many patients are anxious because of the chronic nature of their condition and the apparent inefficacy of their treatments (and, by extension, their doctor). They must be reassured: they are not at risk of blindness, and their disorder should be explained in terms of a gradual loss of the system protecting against environmental aggression. They must be taught to manage their treatment in the long term, according to environmental factors and their own sensations. Too many patients consider their treatment to be ineffective simply because they use their eyedrops at fixed times in the morning and evening, whereas it is during the day that they suffer!
More specific treatments should become available in the near future, especially cyclosporine. If its efficacy is confirmed in humans, it will be a true revolution: at last we will be able to treat the ocular disorder directly instead of using more or less effective substitutive therapy.
References
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


