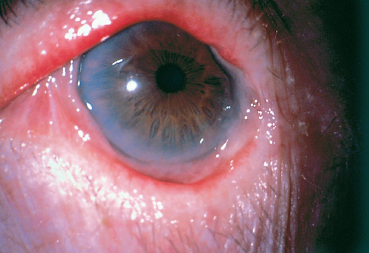
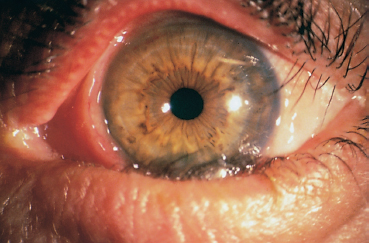
Chapter 4 Fibrosing Conjunctivitis Interactions between fibroblasts and inflammatory cells are responsible for the fibrosis that occurs during the course of chronic conjunctival inflammation. Some causes of fibrosing conjunctivitis are evident, such as trachoma, Stevens-Johnson syndrome, and toxic epidermal necrolysis (Lyell syndrome). Others are more difficult to diagnose and, fortunately, are not always linked to autoimmune conditions like cicatricial pem-phigoid, which is often overdiagnosed. The clinician must know and correctly diagnose the different causes of cicatricial conjunctivitis if the patient is to receive the most appropriate treatment. Recent progress in immunology and immunopathology has greatly improved our understanding of the pathogenesis of autoimmune conjunctivitis. When a patient presents with progressive fibrosing conjunctivitis, it is crucial to confirm the diagnosis of autoimmune conjunctivitis, because treatment involves systemic cytotoxic chemotherapy with potentially severe adverse effects. Autoimmune Fibrosing Conjunctivitis Background Bullous skin diseases have been known since Hippocrates,1 who was the first to use the word pemphigoid when describing different types of fever, including pemphigoides pyretoi, which was probably a febrile bullous condition. The first reports on the association of pemphigoid with conjunctivitis were attributed to Wichmann in 1793,1 and later to Cooper, who called the syndrome “pemphigus of the conjunctiva.”2 In a review of the literature on cicatricial pemphigoid in 1900, Franke underlined the poor prognosis of this sight-threatening form of fibrosing conjunctivitis.3 Many different names have been used for autoimmune fibrosing conjunctivitis, including ocular pemphigus,4 essential shrinkage of the conjunctiva,3 benign mucous membrane pemphigoid,5 and, finally, ocular cicatricial pemphigoid (OCP), which is now the most common term.6 The differentiation and classification of autoimmune bullous skin diseases started in the 1950s. The dermatologists Lever et al. were the first to show that pemphigus and pemphigoid were two distinct conditions, both clinically and histologi-cally.7 In 1968, Carroll and Kuwabara confirmed this distinction in their ultrastructural studies.8 Pemphigus is characterized by intraepidermal bullae secondary to acantholysis, whereas in pemphigoid the blisters are subepidermal with cleavage occurring at the dermal-epidermal junction, within the basement membrane zone (BMZ). It was only in the early 1980s that ophthalmologists realized that fibrosing conjunctivitis could be associated with various autoimmune mucocutaneous diseases.6,9 There is no consensus classification of autoimmune conjunctivitis, and ophthalmologists are currently using the same immunopathological approach as dermatologists. Autoimmune fibrosing conjunctivitis can be isolated or associated with autoimmune conditions affecting the skin and/or other mucosae.6,10 Epidemiology The precise incidence of autoimmune conjunctivitis is unknown. If one considers that the main cause of autoimmune fibrosing conjunctivitis is cicatricial pemphigoid, the incidence of this condition can be estimated at between 1 per 8000 and 1 per 46 000 ophthalmological patients, depending on the author.6,11,12 These Figures probably underestimate the incidence of cicatricial pemphigoid, being based solely on ophthalmologists’ observations and therefore leaving out patients without ocular involvement who are treated by stomatologists, dermatologists, or geriatricians. Another factor in this underestimation is that most of the cases taken into account involve severely affected patients who already have incapacitating fibrosis. The age at onset of autoimmune conjunctivitis depends on the cause. Conjunctivitis associated with bullous pemphigoid classically affects the elderly, although cases have been described in young people. The mean age at diagnosis of cicatricial pemphigoid is 64-70 years,6,10,12 but there have also been well-documented cases in children.13,14,15 Other autoimmune bullous skin conditions generating fibrosing conjunctivitis, such as epidermolysis bullosa acquisita and dermatitis herpetiformis, tend to affect far younger patients. Isolated or pure autoimmune fibrosing conjunctivitis was recognized, too, recently for meaningful epidemiological data to have accumulated. Pathogenesis There are abundant arguments pointing to autoimmunity as the mechanism underlying some forms of conjunctivitis, including coexistence with other autoimmune conditions, the presence of immune deposits in the conjunctiva, detection of circulating au-toantibodies against the BMZ or the intercellular substance of the conjunctival epithelium, and the efficacy of immunomodu-latory and/or immunosuppressive therapies.16–29 The immuno-logical mechanism behind autoimmune conjunctivitis seems to be type II antibody-mediated cytotoxicity.17 Standard histologi-cal examinations, combined with fluorescence and enzymatic immunolabeling techniques, can be used to identify the type of conjunctival inflammatory infiltrate, which is usually mononu-clear and nonspecific.6 Levels of almost all inflammatory mediators have been found to be strongly increased in both the conjunctival epithelium and chorion of patients with cicatricial pemphigoid.6 The cellular composition of the infiltrate can vary according to the cause, activity, and progression of the condition.30,31 These investigatory techniques can of course also be applied to the skin and other mucosae of patients who have associated extraocular involvement. Numerous insults to the conjunctiva, of a traumatic, chemical, inflammatory, or infectious nature, can induce fibrosis. In autoimmune conjunctivitis, and particularly in cicatricial pemphigoid, the fibrosing process is characterized by its inexorable progression in the absence of treatment. The mechanisms responsible for the fibrosis, whatever the underlying cause, are poorly known and multifac-torial, but they do not only involve inflammation. This would explain the progression of fibrosis despite apparent control of the ocular inflammation in some cases. Some of the target antigens in autoimmune conjunctivitis have now been identified. They are represented by epitopes of the numerous molecules composing the junction complex between the epithelium and chorion in subepithelial autoimmune conditions,32–37 and desmosome epitopes in autoimmune intraepithelial diseases.38,39,40 Detection of circulating autoantibodies (whose pathogenic role is poorly known) is rare in cicatricial pemphigoid and epidermolysis bullosa acquisita, but more frequent in bullous pemphigoid.6,34 A genetic predisposition is found in some forms of autoimmune conjunctivitis. In cicatricial pemphigoid, for example, a highly significant statistical link has been found with the DQw7 gene (DQb1 *0301 ).41,42 A link with the HLA-DR4 antigen has also been found in many cases43. HLA-DR2 has been linked to epidermolysis bullosa acquisita and is also more frequent in dark-pigmented patients.44 Clinical Aspects of Conjunctival Fibrosis Conjunctival fibrosis is not specific to autoimmune conjunctivitis, as it occurs in many other settings (see p. 87 ff). Autoimmune conjunctivitis usually affects both eyes. It is associated with variable symptoms of ocular irritation, including a foreign body sensation, a burning sensation, photophobia, lacrimation, pruritus, ocular pain, mucus discharge, and hyper-emia. However, the severity of the disease is not always proportional to the intensity of these symptoms. Progressive fibrosing conjunctivis can be almost asymptomatic in some patients.6,9 The first signs of subepithelial fibrosis mainly affect the inferior and superior tarsal conjunctiva. Initially they consist of fine white lines that are often perivascular (Fig. 4.1). Progression of the fibrosis leads to fornix foreshortening (Fig. 4.2), symblephara (Fig. 4.3), and, in the terminal phase, ankyloblepharon formation (Fig. 4.4). The consequence of this fibrotic process is the destruction of goblet cells, the obliteration of lacrimal gland ductal orifices, and a severe dry eye syndrome (Table 4.1). Conjunctival fibrosis also modifies the architecture of the eyelids and adnexa, producing aberrant eyelash growth and entropion-trichiasis (Fig. 4.5). Fig. 4.1 Stage I conjunctival fibrosis: whitish subconjunctival striae Fig. 4.2 Stage II conjunctival fibrosis: fornix foreshortening Fig. 4.3 Stage III conjunctival fibrosis: symblephara Fig. 4.4 Stage IV conjunctival fibrosis: ankyloblepharon Fig. 4.5 Entropion-trichiasis secondary to conjunctival fibrosis Table 4.1 Consequences of conjunctival fibrosis on the tear film
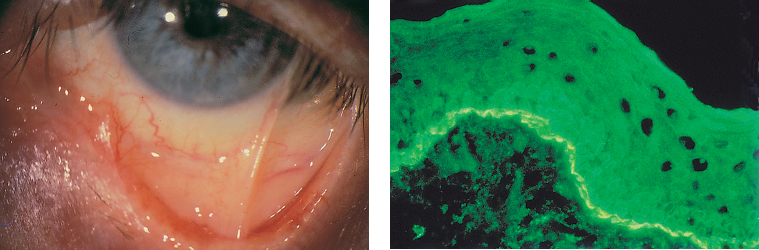
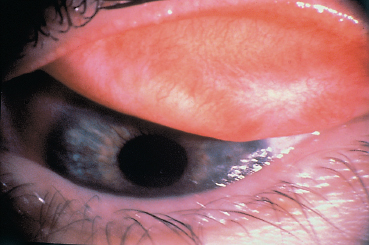
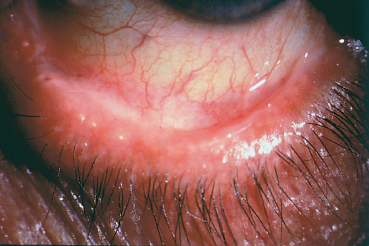
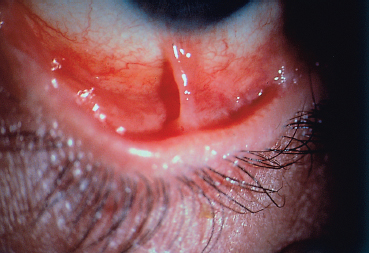
| All the components of the tear film can be affected by conjunctival fibrosis: —The aqueous component: the orifices of the main and accessory lacrimal gland ducts and ductules are obliterated by the fibrotic process; —The mucin component: the goblet cells (which synthesize the mucins forming the deep tear film layer) are damaged by the fibrotic process. Their loss is also induced by the mediators of the associaced inflammation; —The lipid component: secondary meibomitis is often present, and modifies the tear film lipid layer, leading to its instability and increased tear fluid evaporation. |
Corneal complications are favored by dryness, trichiasis, en-tropion, and corneal exposure. They consist of epithelial punctate keratitis and epithelial defects, but they can be more severe, including stromal ulcerations, neovascularized scars, bacterial or fungal superinfection, perforation, and xerosis, and they can lead to corneal blindness11,12 (Fig. 4.6).
These inflammatory diseases of the ocular surface are often complicated by or associated with chronic glaucoma, which may be underdiagnosed, as ophthalmologists sometimes do not measure the intraocular pressure of these eyes with an often irregular corneal surface. The pathogenesis of chronic glaucoma in these settings is unclear; the main hypotheses are genetic predisposition, trabeculitis, adverse effects of corti-costeroid therapy, and a rise in episcleral venous pressure secondary to fibrosis.45
In autoimmune fibrosing conjunctivitis, conjunctival hyper-emia also is important to be assessed. It can be induced by the release of inflammatory mediators including cytokines (tumor necrosis factor [TNF], interleukins), and also histamine. It is a good marker of activity of the inflammatory process, provided there is no associated mechanical irritant such as ectopic eyelashes. It is rarely possible to see “blisters” on the conjunctiva and the corneal surface, because they are very transient. In most cases the ophthalmological examination reveals only post-bullous corneal erosions.6,9
Table 4.2 Classification of conjunctival fibrosis (according to Tauber J et al.46):
| —Stage I: Subconjunctival fibrosis. |
| —Stage II: (Lower) fornix foreshortening. II A: 0-25% II B: 25-50% II C: 50-75% II D: 75-100% |
| —Stage III: Symblephara (% length of lower lid affected). III A: 0-25% III B: 25-50% III C: 50-75% III D: 75-100% The number of symblephara observed at the lower fornix (by pulling the lower lid downward, with the eye looking upward) is added in Arabic characters. |
| —Stage IV: Ankyloblepharon. |
To assess the progression of the fibrotic process and treatment efficacy, it is important to quantify reproducibly both the fibrosis and the conjunctival inflammation (reflected by conjunctival hyperemia). Tauber et al.46 offer a classification of fibrosis that makes an excellent compromise between the systems proposed by Mondino and Foster6,12 (Table 4.2). Mon-dino’s system uses as an index only the degree of inferior fornix foreshortening, while that of Foster takes into account the presence of fornix foreshortening and symblephara, but not their severity. The classification of Tauber et al. is ideally completed by scoring bulbar conjunctival hyperemia on the basis of four quadrants on a scale of 0-4 (Figs. 4.7 and 4.8). Wright et al. have developed another classification that quantifies inflammation and distinguishes three types of cicatricial conjunctivitis—acute, subacute, and chronic.9,47 The acute stage is defined as a highly inflammatory conjunctiva with conjunctival erosions. The subacute stage corresponds to marked conjunctival hyperemia associated with ocular symptoms. The chronic stage is characterized by a white conjunctiva with only subconjunctival fibrosis. None of the available classifications takes into account palpebral and corneal signs.
Laboratory Investigations
The conjunctival biopsy is easily performed under local anesthesia with subconjunctival lidocaine. The sample (mean: 5×7 mm) is harvested from the bulbar conjunctiva adjacent to the limbus, if possible in an inflamed area (Fig. 4.9). Sutures are unnecessary. Biopsy of the fornix is contraindicated because it worsens its shrinkage and stimulates symblepharon formation.6 An antibiotic and antiinflammatory ointment is prescribed for a few days. The sample is divided into as many fragments as are required. Generally, a piece is placed in a fixative such as formol (which allows immunoenzymatic studies on paraffin slices) for histological examination; another piece is placed in normal saline and immediately frozen for immuno-fluorescence and immunoenzymatic studies; a third fragment can be processed for immunoelectron microscopy following sophisticated methods.
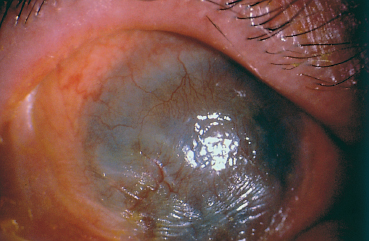
Fig. 4.6 Corneal neovascularization and opacification and xerosis, secondary to conjunctival fibrosis
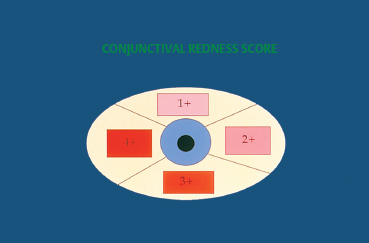
Fig. 4.7 Redness score (0-4)
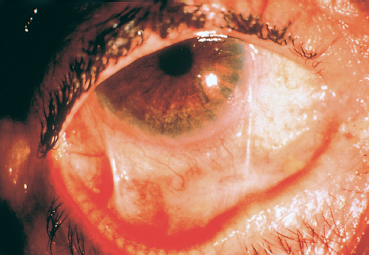
Fig. 4.8 Example of a scoring system for fibrosis and conjunctival hyperemia (modified classification of Tauber et al.). Stage IIC-IIIB2 (right eye) = lower fornix foreshortening (50-75 %) associated with two symblephara occupying 25-50% of the length of the lower lid. The redness score is 2 in the temporal and lower quadrants
Pathology
Histological studies contribute little to the diagnosis of autoimmune fibrosing conjunctivitis because of their poor specificity.6 Contrary to histological examination of cutaneous lesions and extraocular mucosal lesions, the detection of BMZ cleavage in the conjunctiva is of no value for diagnosing autoimmune conjunctivitis because the conjunctiva is very fragile, and any histological epithelial detachment most likely will represent a fixation artifact. Histological findings in OCP include a reduction in the number of goblet cells and squamous metaplasia of the conjunctival epithelium (Fig. 4.10). The chorion shows nonspecific chronic inflammation with a cellular infiltrate consisting of lymphoplasmocytic mononuclear cells and, in some cases, perivasculitis. Mast cell numbers are often increased, and eosinophils and neutrophils are sometimes found. The latter can be numerous during the acute phase of the condition, particularly in the superficial chorion. Fibrosis secondary to collagen neosynthesis by fibroblasts also can be observed.6,48,49
Transmission electron microscopy studies have clearly characterized the conjunctival lesions of OCP.50 The epithelium contains markedly increased numbers of desmosomes and an increased density of intracytoplasmic tonofilaments and tono-fibrils. This is accompanied by a rarefaction of goblet cells. The BMZ is discontinuous, duplicated, and thickened in places. The chorion contains a disorganized inflammatory infiltrate consisting of cells, fibrin, and collagen.
Scanning electron microscopy shows large homogeneous deposits of amorphous material resembling thick mucus at the conjunctival surface.51
Direct Immunolabeling
Direct Immunofluorescence6,52,53
The chorioepithelial junction of the conjunctiva was long considered as a simple linear amorphous structure called BMZ that could only be observed by light microscopy after special staining such as periodic acid-Schiff (PAS). It is in fact a morphologically and biochemically complex structure involved in various biological and pathological processes, including autoimmune subepithelial conjunctivitis. Direct immunofluorescence is the oldest and simplest immunolabeling technique to reveal immune deposits composed of autoantibodies fixed to their target antigens at the level of the BMZ (Fig. 4.11). It is simpler when applied to biopsy specimens of cutaneous or buccal lesions. The antibodies used to detect these deposits are poly-clonal antibodies coupled to fluorescein and sometimes to rho-damine. They are directed against human immunoglobulin (especially IgG, IgA, and IgM) or the corresponding Fc fragment, and against some complement factors (C3 and C4). A typical continuous linear fluorescent deposit along the epithelial BMZ characterizes autoimmune mucocutaneous cicatricial diseases (Fig. 4.12). However, direct immunofluorescence cannot differentiate the various autoimmune sub-epithelial conditions.
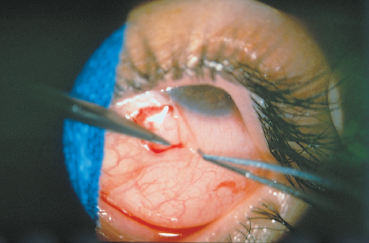
Fig. 4.9 Conjunctival biopsy
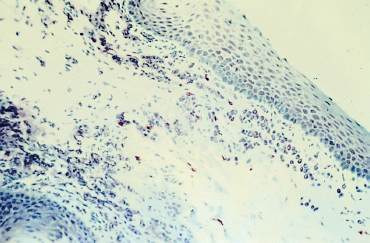
Fig. 4.10 Ocular cicatricial pemphigoid: histological section of the conjunctiva. Note the squamous metaplasia, loss of goblet cells, and inflammatory cell infiltration of the chorion (Giemsa staining, original magnification x 40)
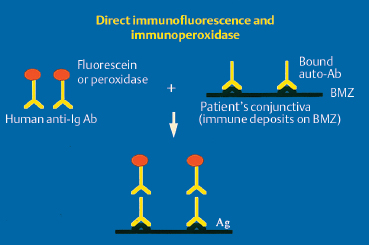
Fig. 4.11 Principle of direct immunofluorescence and immunoperoxidase labeling.
Ab: antibody; Ag: antigen; Ig: immunoglobulin; BMZ: basement membrane zone
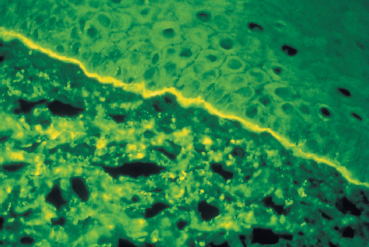
Fig. 4.12 Ocular cicatricial pemphigoid: immune deposits form a continuous line of fluorescence along the conjunctival BMZ
In autoimmune intraepithelial conjunctivitis (pemphigus), immune deposits are detected at the intercellular junctions and give a fishnet immunofluorescent pattern.8
Immunoenzymatic Techniques
The principle of these methods is identical to that of direct immuno-fluorescence, except that the antibodies are not coupled to fluorescein but rather to peroxidase (Fig. 4.11). The reaction of peroxidase with avidin biotin stains the immune deposits brown. Immunoenzymatic labeling, which is a little more complex and costly than direct immunofluorescence, is far more sensitive for the diagnosis of autoimmune conjunctivitis6,54,55 (Fig. 4.14).
The use of immunoperoxidase also permits more thorough immunopathological studies with monoclonal antibodies directed against cell surface antigens (CD antigens) and cytokines.6,31 Thus, studies of patients with OCP showed that inflammatory cells colonizing the conjunctival epithelium were T helper cells (CD4+), dendritic cells (CD1+), and macrophages (CD14+), and that the T helper/T suppressor ratio was significantly higher than in the normal conjunctiva. The dense inflammatory cell population infiltrating the conjunctival chorion of the same patients is composed of T lymphocytes (CD3+ and CD5+), including T helper (CD4+) and T suppressor cells (CD8+), macrophages (CD14+, MAC1+), and dendritic cells (CD1+ and HLADR+). Ten percent of these cells express IL-2 receptors (CD25+) at their surface, reflecting their activation. The composition of the subepithelial inflammatory cellular infiltrate varies with the activity of the condition. Macrophages and polymorphonuclear neutrophils are abundant in the acute phase. T lymphocytes predominate in subacute forms; CD8+ cells outnumber CD4+ cells, whereas they are present in equal numbers in the acute phase.31 A costimulatory signal deriving from antigen-presenting cells seems necessary to maintain the activation of T helper cells. Over-expression of costimulatory B7-2 ligands (CD86) has also been demonstrated in the conjunctival chorion of patients with OCP.55a
Direct Immunoelectron Microscopy
Direct immunoelectron microscopy is the most sophisticated technique for demonstrating the presence of autoantibodies fixed to target autoantigens in situ. The main difference with direct immunofluorescence is the choice of markers, which must be electron-opaque for transmission electron microscopy. These markers are peroxidase and colloidal gold (immunogold). Immune deposits can be detected within the epithelium in autoimmune intraepithelial conjunctivitis and in the different areas of the epithelial BMZ in autoimmune sub-epithelial conjunctivitis. They appear as dark electron-dense formations56–59 (Figs. 4.15 and 4.16).
The ultrastructure of the BMZ, where the adhesion complex between the epithelium and chorion is located, is described in another chapter (see p. 18 ff). It comprises, from top to bottom, the hemidesmosomes, the BMZ itself (composed of the lamina lucida and lamina densa, and crossed by anchoring filaments) and the zone of anchoring fibrils.
Ultrastructural localization of the immunolabeled target antigens (in the lamina lucida, lamina densa and anchoring fibril zone) can thus help to determine more precisely the nature of the autoimmune condition. Immunoelectron microscopy, already used in dermatology, is being assessed for its diagnostic sensitivity in ophthalmology.58,59
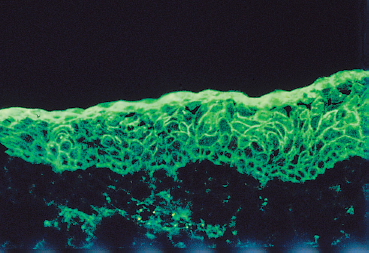
Fig. 4.13 Pemphigus: characteristic fishnet immunofluorescent pattern of the epithelial intercellular substance
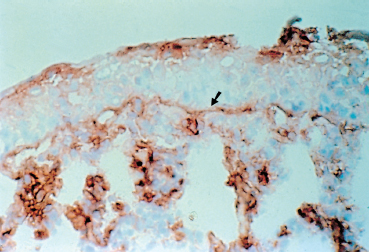
Fig. 4.14 Immune deposits (IgA) form a continuous brown line along the conjunctival BMZ (arrow). Also note the labeling of the epithelial surface (IgA derived from tears) and of inflammatory cells in the chorion
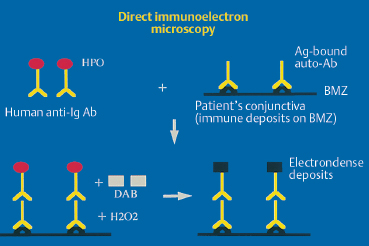
Fig. 4.15 Principle of direct immunoeletron microscopy.
Ab: antibody; Ag: antigen; Ig: immunoglobulin; BMZ: basement membrane zone; HPO: horseradish peroxidase; DAB: diaminobenzidine
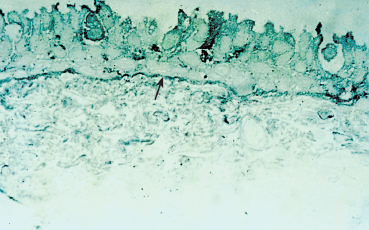
Fig. 4.16 Semi-thin section: linear immune deposits on the BMZ (arrow). Ultrastructural studies show that the deposits are located within the BMZ (see Figs. 4.17 and 4.18)
Circulating Antibodies
The detection and titration of circulating autoantibodies directed against the BMZ and epithelial intercellular substance are based on techniques that can now be used in many immunology laboratories. These methods are based on reacting the patient’s serum with a cutaneous or mucosal substrate (most target antigens in the conjunctiva are apparently identical to those in other mucosae and the skin). Autoantibodies fixed to the target antigen are visualized on the substrate by means of a “sandwich” technique with labeled antihuman immunoglobulin antibodies.
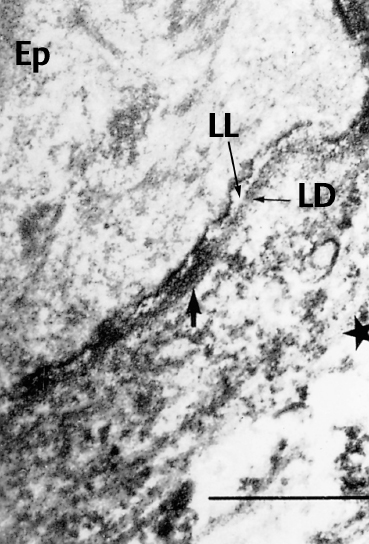
Fig. 4.17 Direct immunoelectron microscopy of OCP: the electrondense immune deposits (arrows) are located both on the lamina lucida (LL) and the lamina densa (LD) of the conjunctival BMZ (original magnification x 12 000).
Ep: epithelium; *: chorion
Indirect Immunofluorescence
In indirect immunofluorescence, the antihuman immuno-globulin antibodies used to detect substrate-bound autoantibodies are coupled to fluorescein. The substrates most commonly used are salt-split or normal human skin and rat esophageal mucosa (Fig. 4.19).6,18,25,26,27 The use of salt-split skin can sometimes help to orient the diagnosis of bullous sub-epithelial autoimmune conditions, especially by distinguishing between bullous pemphigoid and epidermolysis bullosa acquisita. The fluorescence classically stains the roof of the cleavage in the former and the floor in the latter.60 In contrast, in cicatricial pemphigoid, the location of fluorescence, when present (which is rare), is not clearly established.37 The conjunctiva has also been tested as a substrate for autoimmune conjunctivitis and appears to be more suitable.52
Western Immunoblotting and Immunoprecipitation
These two tests can be used to precisely characterize the target antigens of circulating autoantibodies on the basis of their molecular weight. The diversity of substrates explains the conflicting flicting results obtained in different studies. These substrates can be epidermal and dermal extracts,32,33,63,64,65 or other extracts of cutaneous or conjunctival origin.35,61,62 They are placed in contact with the serum to be tested and submitted to electro-phoretic migration in polyacrylamide gel containing sodium dodecyl sulfate (SDS). Once separated, the proteins are transferred to a nitrocellulose sheet and stained. The protein bands corresponding to the antigen-autoantibody complexes are compared with reference bands to determine the molecular weight of the target antigens (Fig. 4.21). Some results also support the notion that isolated autoimmune conjunctivitis is a distinct entity.33,63,65a Recently, molecular biology techniques were used to characterize some BMZ proteins and to clone the hap-tens responsible for mucocutaneous autoimmune diseases.66
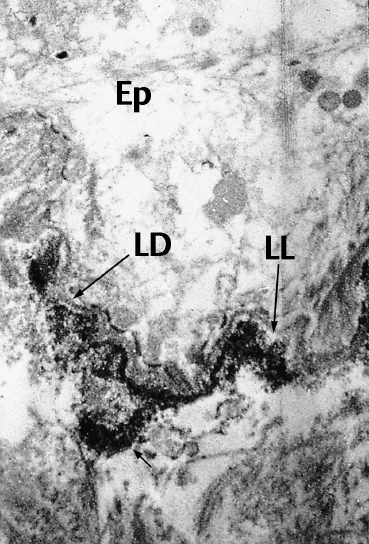
Fig. 4.18 Direct immunoelectron microscopy of epidermolysis bullosa acquisita: the electrondense immune deposits (arrows) are located under the LD in the region of the anchoring fibrils (original magnification x 10 000)
Ep: epithelium; *: chorion;
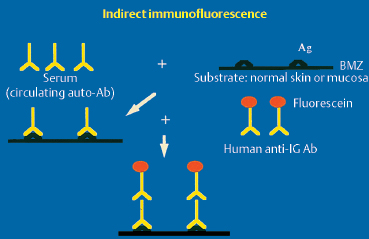
Fig. 4.19 Principle of indirect immunofluorescence
Ac: antibody; Ag: antigen; Ig: immunoglobulin; BMZ: basement membrane zone
Indirect Immunoelectron Microscopy
Indirect immunoelectron microscopy has so far only been used in dermatological research to characterize circulating antibodies in autoimmune bullous dermatosis (AIBD). The method is based on both indirect immunofluorescence and direct immunoelectron microscopy. The serum to be tested is incubated with normal skin obtained during plastic surgery. The mixture is placed in contact, in a sandwich approach, with antibodies directed against human immunoglobulin heavy chains labeled with horseradish peroxidase. After incubation with diaminobenzidine, electron microscopic examination shows the antigenic targets of circulating autoantibodies (Fig. 4.20). It is even possible, thanks to various technical variants (e.g., saponin), to reveal intracellular antigens.37 These advanced research techniques are highly promising.
Causes
Conjunctivitis Associated with Autoimmune Bullous Dermatoses (Tables 4.3, 4.4)
Autoimmune Diseases of the Basement Membrane Zone
General considerations
Autoimmune conjunctivitis is a rare condition. It usually is associated with a condition affecting the skin and other mu-cosae, known as autoimmune bullous dermatosis (AIBD). A classification of AIBD has been established on the basis of clinical criteria, including the topography of the bullae or other skin lesions, the type of progression, scarring, and the presence of mucosal lesions (Tables 4.5 and 4.6).67 These latter mainly involve the buccal mucosa. The nasal, pharyngolaryngeal, esophageal, anal, and genital mucosae are far more rarely affected. The nosological classification of these entities has progressed thanks to new immunological, immunopathological, and molecular biology methods (Table 4.4).
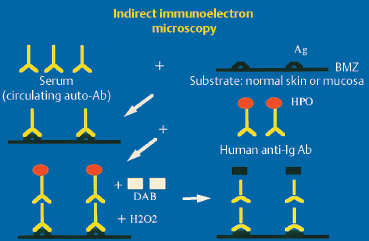
Fig. 4.20 Principle of indirect immunoelectron microscopy
Ab: antibody; Ag: antigen; Ig: immunoglobulin; BMZ: basement membrane zone; HPO: horseradish peroxidase; DAB: diaminobenzidine
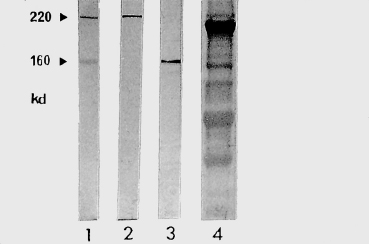
Fig. 4.21 Western immunoblot (courtesy of Dr. C. Prost):
1: Bullous pemphigoid (BP): the 220-kDa and 160-kDa antigens correspond to antigens BP 230 and BP 180; 2: BP; 3: BP or cicatricial pemphigoid; 4: Control
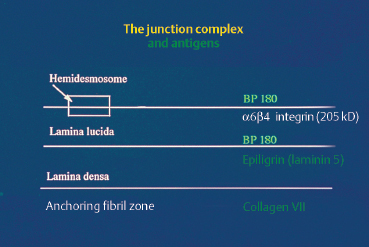
Fig. 4.22 Antigens of the chorioepithelial or dermal-epidermal junction complex
Ocular involvement is associated with major morbidity because it causes fibrotic scars that can lead to corneal blindness.6,9,10,11 The conjunctivitis associated with AIBD is not, however, specific, its clinical characteristics alone being insufficient to diagnose precisely the underlying autoimmune disease. Diagnosis of autoimmune fibrosing conjunctivitis can be difficult when the conjunctivitis appears to be isolated. It can precede by several months or years the involvement of the skin or other mucosae. All available clinical and laboratory tools must be used to identify the cause, as this determines the therapeutic strategies.
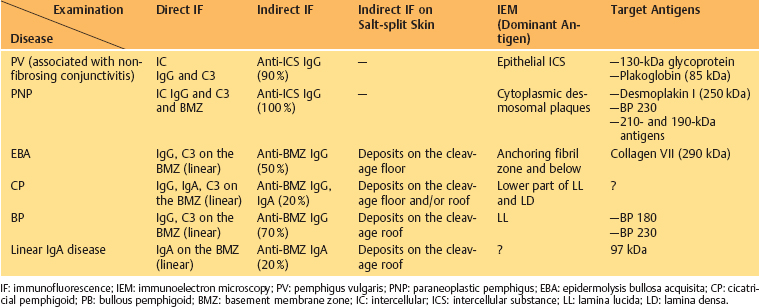
Table 4.3 Autoimmune mucocutaneous diseases (Those associated with conjunctivitis are shown in bold)
| Intraepithelial Autoimmune Diseases: | Subepithelial Autoimmune Diseases: |
|---|---|
| —Pemphigus vulgaris (nonfibrosing conjunctivitis) —Pemphigus vegetans —Pemphigus seborrheic —Pemphigus foliaceus —Pemphigus erythematosus —Brazilian pemphigus —Paraneoplastic pemphigus | —Bullous pemphigoid —Cicatricial pemphigoid —Herpes gestationis —Epidermolysis bullosa acquisita —Bullous systemic lupus erythematosus —Linear IgA disease —Dermatitis herpetiformis —Lichen planus pemphigoides —(Paraneoplastic) lichen planus |
Table 4.5 Cutaneous manifestations of autoimmune subepithelial mucodermatoses
| —Cicatricial pemphigoid: * Adults * Scarring * Prevalence of lesions on head and trunk * Tender bullae (inconstant) * Mucosal involvement (very frequent) |
| —Epidermolysis bullosa acquisita: * Adults * Bullae on extensor surfaces * Fragile skin (traumatic bullae) * Resistance to corticosteroids * Formation of milia * Mucosal involvement and positive Nikolsky sign (inconstant) |
| —Bullous pemphigoid: * Adults * Tense bullae * Peribullous skin involvement (no bullae on healthy skin) * Mucosal involvement (generally rare) * Nikolsky sign negative on healthy skin |
| —Linear IgA disease: * Adults and children * Tense bullae with herpetiform clusters of vesicles * On erythematous or normal skin * Often lower trunk, perineum, thighs; sometimes scalp * Mucosal involvement (possible) |
| —Dermatitis herpetiformis: * Adults * Tender bullae with herpetiform clusters of vesicles * Elbows, knees, shoulders, sacral region, scalp * Mucosal involvement rare * Gluten-sensitive enteropathy |
Cicatricial pemphigoid
The terms variously used to designate cicatricial pemphigoid include “benign mucous membrane pemphigoid,” “ocular pemphigus,” “desquamative gingivitis,” and “localized cicatricial pemphigoid.” Cicatricial pemphigoid is an autoimmune subepithelial disease of the elderly (mean age: 55-70 years) that mainly affects the buccal mucosa (85%) (Figs. 4.23 and 4.24), the conjunctiva (65%) (Figs. 4.1–4.4, 4.28), and, more rarely, the skin (25%) (Fig. 4.25). Some forms remain localized to a single mucous membrane, such as pure OCP and isolated desquamative gingivitis. Cicatricial pemphigoid is rarely life-threatening, contrary to bullous pemphigoid.
The cutaneous blisters are typically tense and located on erythematous or healthy skin. They are limited to the upper part of the body, i.e., the neck, face, and scalp. The skin lesions are poorly specific, however, and diagnostic problems can arise with other forms of AIBD.67 Localized cicatricial pemphigoid (Brunsting-Perry type) is the pure cutaneous form of cicatricial pemphigoid.
Table 4.6 Diagnostic criteria for autoimmune subepithelial fibrosing conjunctivitis
| * Conjunctival fibrosis —Symptomatic or asymptomatic, —Inflammatory or noninflammatory, —Progressive or nonprogressive. |
| * Associated with one of the following signs: 1) Immune deposits at the conjunctival BMZ (immunofluorescence or immunoperoxidase [conventional or immunoelectron microscopy] labeling). 2) Characteristic autoimmune bullous dermatitis or immune deposits at the dermal-epidermal junction, or chorio-epithelial junction in an extraocular mucous membrane. 3) Favorable outcome (nonprogression of fibrosis and resolution of conjunctival inflammation) on immunomodulatory and/or immunosuppressive treatment. |
* AND |
The diagnosis of cicatricial pemphigoid is mainly based on immunopathological findings showing continuous linear immune deposits on the BMZ in situ.6,68 Detection of circulating autoantibodies by means of indirect immunofluorescence and Western immunoblotting is inconsistent.6
Immunoelectron microscopy can reveal immune deposits corresponding to the site(s) of the target antigen(s) in the skin and mucosae, including the conjunctiva. They are extracellular, granular, and discontinuous, usually located at the lower part of the lamina lucida, and/or on the lamina densa, obscuring this latter (Fig. 4.17).37,57,58,59,69
The target antigens in cicatricial pemphigoid are unknown, and the results of work in this field tend to conflict (Fig. 4.22). Differences in immunoblotting techniques, especially the substrates used, are largely responsible for this situation. Bernard et al.34,35,70 described a series of eight patients with cicatricial pemphigoid, of whom five had ocular involvement and circulating antibodies recognizing epidermal antigens of 180 kDa and 230 kDa. These molecular weights being identical to those of the antigens involved in bullous pemphigoid (BP180 and BP 230), the authors concluded that immunoblotting was unable to distinguish between the two conditions. In another study of 10 patients with cicatricial pemphigoid and ocular involvement, Mohimen et al., using extracts of COLO and SCaBER tumor cell lysates, identified target antigens of 230 kDa, 160 kDa (in fact, BP180 and BP230), and 205 kDa32; the latter appears to correspond to the protein (34 component of integrin a6|34.66,70a Epiligrin, another component of the dermal-epidermal and chorioepithelial junctions, is also suspected of being the autoantigen in some cases of cicatricial pemphigoid with conjunctival involvement.36,71,72 It is possible that autoantibodies to several components of the BMZ could generate the same clinical picture, i.e., cicatricial pemphigoid.
The participation of eosinophils and their granule proteins (eosinophilic cationic protein [ECP] and major basic protein [MBP]) in tissue damage in the acute phase of inflammation in OCP was discussed.72a The costimulatory molecule CD28 and its ligand B7-2 also may contribute to the sustained immune activation in OCP conjunctiva.72b
Bullous pemphigoid
Bullous pemphigoid is not a rare disease, with an estimated 350 new cases yearly in France alone. It is characterized by spontaneous occurrence in the elderly (mean age: 75 years) of large, tense cutaneous blisters developing on an erythematous base. The blisters predominate on the inner aspects of the thighs, the flexor surfaces of the forearms, the axillae, the groins, the lower abdomen, and the palmar and plantar surfaces. These parts of the body possibly contain a higher density of target antigens. The Nikolsky sign (a dislodging of the epidermidis with lateral finger pressure) is negative. Classically, mucosal lesions are rare and their presence defines atypical bullous pemphigoid. The course is chronic with exacerbations, sometimes induced by drugs. The main characteristic of the condition is that the lesions heal without scarring. Ten percent of cases of bullous pemphigoid appear to be of paraneoplastic origin.67
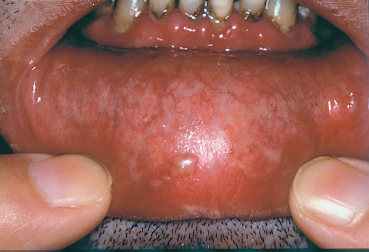
Fig. 4.23 Cicatricial pemphigoid: buccal blisters
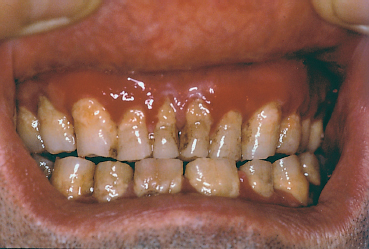
Fig. 4.24 Cicatricial pemphigoid: desquamative gingivitis
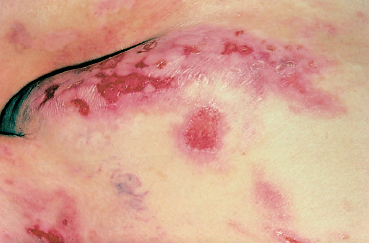
Fig. 4.25 Cicatricial pemphigoid: skin lesions (courtesy of Dr. C. Prost)
Conjunctival involvement in bullous pemphigoid usually consists of mild conjunctivitis, but cases of fibrosis identical to that observed in OCP have been reported.9 Among the laboratory examinations used to confirm the diagnosis of bullous pemphigoid, immunoblotting is the test of choice because it reveals the presence of antigens BP180 and BP230 using an epidermal extract.35 Detection of these antigens is very useful for the diagnosis of “atypical” bullous pemphigoid, especially forms associated with ear, nose, and throat (ENT) mucosal and conjunctival involvement.
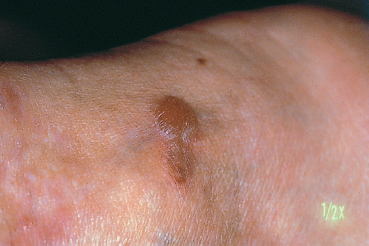
Fig. 4.26 Linear IgA dermatosis: cutaneous blister
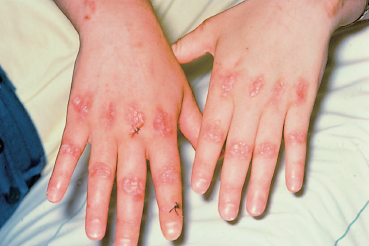
Fig. 4.27 Epidermolysis bullosa acquisita: formation of milia on the hands and fingers (courtesy of Dr. C. Prost)
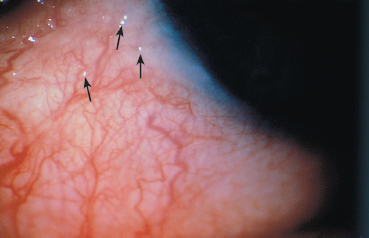
Fig. 4.28 Early-stage OCP: conjunctival microbullae
Direct and indirect immunoelectron microscopy have been used to locate the antigen(s) of bullous pemphigoid, in the form of continuous linear deposits. They are strictly localized at the upper part of the lamina lucida, at the level of the hemidesmosomes of the basal keratinocytes.37,69
Epidermolysis bullosa acquisita
Epidermolysis bullosa acquisita (EBA) is an autoimmune sub-epithelial disease with two recognized clinical forms. The chronic form is rare and clinically resembles hereditary dystrophic epidermolysis bullosa. Onset occurs in adulthood (40-50 years). The blisters occur on normal-appearing skin and can be provoked by a mild trauma. The lesions predominate in areas of friction (joints, extensor surface of the legs, hands, and feet). Mucosal involvement is frequent. The course is chronic and healing is slow with atrophic skin scarring and especially milia (Fig. 4.27). The inflammatory form (inflammatory EBA) can simulate bullous pemphigoid by the aspect of the subcutaneous blisters and the absence of scars and milia formation during initial exacerbations. This form of EBA appears to be frequent and may represent 10-25% of autoimmune sub-epidermal bullous conditions.67
Mucosal lesions are frequent in EBA and their scars resemble those of cicatricial pemphigoid. Conjunctival fibrosis can lead to symblepharon formation and a decrease in visual acuity. Milia are sometimes visible on the eyelids. The ocular involvement can be milder, combining nonfibrosing conjunctivitis with peripheral ulceration and subepithelial blebs on the cornea, possibly reflecting immune conflict within the cornea.73,74,75
Other diseases are associated with EBA in 20-50% of cases, the most frequent being inflammatory bowel diseases and diabetes. Cases of cryoglobulinemia, thyroiditis, rheumatoid arthritis, and Waldenström disease have also been reported.67
The target antigen in EBA is collagen VII. The dermal target antigen is a 290-kDa protein recognized by monoclonal antibody LH 7.2 (Fig. 4.22). The location of this antigen explains why immunoelectron microscopy shows deposits of IgG, IgA, and complement situated in the subbasal lamina-anchoring fibril zone. This ultrastructural feature has been demonstrated in skin and more recently in the conjunctiva (Fig. 4.18).75–78
Linear IgA dermatosis
Linear IgA dermatosis is characterized by cutaneous blisters that disappear without scarring (Fig. 4.26). Conjunctival involvement is frequent (50% of cases) and clinically indistinguishable from that of cicatricial pemphigoid. Some authors consider that linear IgA dermatosis is not a clinical entity because of its clinical polymorphism. In contrast, direct immuno-fluorescence always shows homogeneous linear IgA deposits at the dermal-epidermal junction. The conjunctivitis associated with this skin disease has the peculiarity of sometimes being asymptomatic, even in case of severe fibrosis.79–83
Dermatitis herpetiformis
Dermatitis herpetiformis is a bullous skin disease that is more frequent in HLA-B8-positive subjects. Clinically, the cutaneous blisters and bullae are accompanied by pruriginous erythema. Although mucosal involvement is rare, authentic cases of dermatitis herpetiformis have been reported with fibrosing conjunctivitis mimicking that of OCP. Associated gluten-sensitive enteropathy is almost always present. Immunopathological studies show IgA deposits in the papillae of the superficial dermis.84,85,86
Other autoimmune diseases of the basement membrane zone
—Herpes gestationis (pemphigoid of the pregnant woman)
—Bullous systemic lupus erythematosus
Conjunctival lesions are rare in these two conditions but the diagnosis is generally simple, based on clinical findings and laboratory tests (direct immunofluorescence, anti-nuclear, and anti-DNA antibody tests).67
Autoimmune Intraepithelial Diseases: Pemphigus
Pemphigus refers to a group of autoimmune bullous diseases that affect the skin and mucosae and share certain histological and immunopathological characteristics. Pemphigus can occur at all ages, but mainly in subjects over 50. Typically, the lesions occur on the buccal mucosa before appearing on the skin. The cutaneous bullae are flaccid, occur on healthy skin, are not preceded by pruritus, and progress by exacerbations. The Nikolsky sign is usually positive. The patient often complains of systemic symptoms such as fatigue and malaise. Histological studies show suprabasal epithelial acantholysis and a mono-nuclear inflammatory infiltrate. Direct immunofluorescence shows intercellular immunoglobulin and/or complement deposits, giving the epithelium a highly characteristic fishnet appearance (Fig. 4.13). Conjunctival biopsy is useful when biopsy of the skin and buccal mucosa is noncontributory. The bullous lesions are intraepithelial in the conjunctiva and intraepidermal in the skin. Indirect immunofluorescence detection of intercellular substance-reactive circulating antibodies is very frequent in all forms of pemphigus. The serum level of these autoantibodies appears to be a good index of the activity of the disease.67 Studies of target antigens and their correlation with clinical features have defined several forms of pemphigus. Those associated with autoimmune conjunctivitis are pemphigus vulgaris and, especially, paraneoplastic pemphigus.
Pemphigus vulgaris is characterized by the production of autoantibodies directed against a complex of 210 kDa formed by plakoglobin (a desmosome protein) and a 130-kDa antigen. The conjunctivitis usually does not induce fibrosis. In pemphigus foliaceus the autoantibodies are directed against desmoglein, a desmosomal protein of 160 kDa.39
Paraneoplastic pemphigus was identified more recently,87,87 a and many cases have now been reported in the literature. Autoantibodies are directed against an antigen complex composed of desmoplakins I and II, the BP230 antigen of bullous pemphigoid, and an unidentified epithelial antigen of 190 kDa. Diagnosis of pemphigus paraneoplastic is based on Anhalt’s criteria (Table 4.7).87b The most frequently associated cancers are nonHodgkin’s lymphoma and lung cancer. Mucosal lesions are almost always present and consist of painful buccal erosions and papillary conjunctivitis with hyperemia, ulceration, and possible progression to fibrosis.87a A case of pseudomem-branous conjunctivitis has also been reported.87
Lichen Planus
Lichen planus is a presumed autoimmune condition of unknown cause that usually affects the skin and genital and oral mucosae, with occasional conjunctival involvement. It affects men and women of all races aged 30-60 years. It is estimated to account for 1.4% and 5% of visits to dermatologists and stomatologists, respectively.
The skin lesions are papular, squamous, and violaceous, and are typically located on the limbs and external genitalia. The most characteristic mucosal lesions are painful chronic erosions. The most frequent form is erosive gingivitis, but linear subepithelial fibrosis can also be observed.
The conjunctival inflammation associated with lichen planus is rare but severe, with possible scarring.87c Rare cases of cicatrizing conjunctivitis associated with paraneoplastic lichen planus also were reported.88 However, scarring can be caused by other conditions associated with conjunctival lichen planus. Bullous pemphigoid (lichen planus pemphigo-ides)88a,88b and lupus erythematosus, which are also accompanied by fibrosing conjunctivitis, have been found in association with lichen planus.
The immunohistopathological and ultrastructural characteristics of the conjunctivitis associated with lichen planus are well documented. They are similar to those observed in the buccal mucosa and consist of fragmentation and duplication of the BMZ, with fibrin and fibrinogen deposits but no immunoglobulin or complement factors.88c
Table 4.7 Anhalt’s criteria for the diagnosis of paraneoplastic pemphigus87b
| 1) Mucosal erosions and cutaneous bullae associated with cancer; 2) Intraepidermal acantholysis and keratinocyte necrosis; 3) IgG and complement deposits both in the epidermal intercellular space and on the BMZ (dermal-epidermal junction); 4) Circulating autoantibodies directed against the simple and complex epithelia; 5) Serum autoantibodies recognizing (by immunoprecipitation) desmoplakin I (250 kDa), desmoplakin II (210 kDa), BP 230 antigen (230 kDa), and a 190-kDa protein. |
The standard treatment of cutaneous or buccal lichen planus is local and/or systemic corticosteroid therapy. In case of refractory oral lesions or poor tolerance of steroid therapy, cyclosporine mouthwash solution has proved effective. Cyclo-sporine 2% eyedrops have also been successful in conjunctival lichen planus. Cyclosporine acts by inhibiting helper T cells and by modulating the IL-2-dependent immune cascade.
Pure Ocular Cicatricial Pemphigoid
By definition, this is a form of autoimmune fibrosing conjunctivitis with no cutaneous or extraocular mucosal involvement. The diagnosis of autoimmunity is based on direct immunopathological detection of immune deposits in the conjunctival BMZ. This form of conjunctivitis is considered by some as a clinical and immunopathological variant of cicatricial pemphigoid. One study showed IgA autoantibodies directed against a 45-kDa antigen present in keratinocyte culture extracts.33 Other authors have not detected circulating autoantibodies in patients with isolated autoimmune conjunctivitis.63 The results of one direct immunoelectron microscopic study supported the hypothesis whereby pure OCP is characterized by target antigens different from those involved in cutaneous and mucosal cicatricial pemphigoid.89 The results of this study showed that immune deposits were strictly situated within the lamina lucida below hemidesmosomes, while in OCP associated with cutaneous and mucosal involvement they were located in the lower part of the lamina lucida and on the lamina densa.
Presumed Drug-Induced Ocular Cicatricial Pemphigoid
Drugs have been incriminated in the onset of fibrosing conjunctivitis with clinical features almost identical to those of OCP. Most cases involve chronic use of eyedrop preparations containing pilocarpine, epinephrine, dipivephrine, timolol, idoxuridine, ecothiopate, and demecarium. An oral beta-blocker, practolol, has also been incriminated. Several hypotheses have been forwarded to explain these cases of presumed drug-induced OCP. The association may be purely coincidental, the beginning of treatment coinciding with the onset of autoimmune fibrosing conjunctivitis. The fibrosis may also be linked to direct toxicity of the drug or its preservative (see Chapter 8). The fact that the condition in some patients stabilizes on drug withdrawal supports this latter hypothesis. Another explanation is that some drugs might trigger or accelerate the onset of true autoimmune conjunctivitis in predisposed patients.89a This hypothesis is supported by the existence of cases of strictly unilateral proved autoimmune conjunctivitis affecting only the eye to which the eyedrops (generally antiglaucoma or antiherpetic preparations) were applied. Presumed drug-induced OCP could thus be added to the list of drug-induced autoimmune conditions such as disseminated lupus erythematosus, some forms of autoimmune hepatitis, and bullous pemphigoid secondary to the use of some der-matological topics. However, follow-up of patients with unilateral presumed drug-induced OCP has shown that the other untreated and initially unaffected eye sometimes develops fibrosing conjunctivitis. It is thus best to use the term “eyedrop-induced fibrosing conjunctivitis,” which makes no presumptions on what are probably the multiple mechanisms underlying the fibrosis in this setting.6,90
Therapy
The management of autoimmune fibrosing conjunctivitis requires an excellent knowledge of systemic antiinflammatory and immunosuppressive therapies. Autoimmune conjunctivi-tides, such as OCP, are systemic conditions in which an immunological disturbance leads to the production of autoanti-bodies that bind to the conjunctiva. As a rule, systemic diseases require systemic therapy, and local therapy alone cannot halt the inexorable aggravation of these autoimmune conditions. But immunosuppressive therapy cannot be undertaken without close collaboration between the ophthalmologist and an internist, oncologist or experienced general practitioner. Indeed, the medications are prescribed for long periods and life-threatening adverse effects can occur if the drugs are incorrectly used. The risks inherent in these aggressive treatments also call for a full work-up to confirm the diagnosis. Im-munopathological examination of conjunctival biopsy specimens is crucial. The choice of therapy will depend on the patient’s general health and age, and the prognosis of the disease. Despite control of the inflammatory process, further visual loss occurs in severe forms of the disease.90a
Surgery is frequently required to treat conjunctival and palpe-bral scars and to restore visual transparency. It must ideally be performed only when the underlying condition has been controlled by medical treatment, to prevent postoperative inflammatory flare-up. One exception to this rule is the existence of a mechanical irritant, such as ectopic eyelashes, which must be removed surgically as it otherwise maintains the inflammation and hinders the assessment of endogenous inflammation.
Medical Therapy
Drugs
The modern history of treatment of autoimmune fibrosing conjunctivitis began in 1974, when Dave and Vickers first reported the efficacy of systemic cytotoxic immunosuppressive therapy with azathioprine combined with systemic corticosteroids in OCP.91 Later, successful results with cyclo-phosphamide also have been obtained in this setting.92 Beneficial casuistic experiences also were reported with mycophenolate mofetil in some ocular immunological disorders, including OCP.92a Unfortunately, in some cases the inflammatory process resists to immunosuppressive drugs, even when they are combined with high-dose steroids.93 Recently, intravenous immunoglobulin immunomodulatory therapy was tested successfully in treatment-resistant OCP.93a,93b
Noncytotoxic drugs are available for the treatment of mild to moderate forms of autoimmune fibrosing conjunctivitis, and to be used as sparing agents. In 1982, Rogers showed that dap-sone, a sulfone initially prescribed in leprosy and then in dermatitis herpetiformis, relapsing polychondritis and bullous pemphigoid, could be used to treat cicatricial pemphigoid.94 Sulphapyridine, an oral sulfonamide with immunosuppressive and antiinflammatory properties, also proved effective in about 50% of cases in a series of patients with OCP, and is a good alternative to dapsone.95 Sulfasalazine, the precursor of sulfapyridine, also can be used.95a Methotrexate and systemic and topical cyclosporine A,29 have also been tried in this setting but with unconvincing results. Ramzy et al. published an exhaustive review of the modes of action, indications, and adverse effects of immunomodulatory and immunosuppressive drugs in ophthalmology.96
Topical and subconjunctival treatments are not effective in autoimmune conjunctivitis. On the contrary, besides the well-known cataractogenic and glaucomagenic effects of topical steroid therapy, long-term therapy with eyedrops containing preservatives may induce conjunctival fibrosis (see Chapter 8). In fact the only useful topical treatments are the tear substitutives and antibiotics for superinfections. Few trials of subconjunctival mitomycin C injections have shown promising results, but these need to be confirmed.97,97a
Indications
Ocular cicatricial pemphigoid
It is difficult to formulate a standard treatment for OCP. Published results on this rare condition are inadequate, follow-up is generally too short, long periods of spontaneous remission occur in a third of patients, and the relapse rate is high (22%) on treatment cessation.98 The largest series so far involved 104 patients with a mean follow-up of 4 years.92,98 Furthermore, while sex and age have no significant influence on outcome, this is not the case in the stage of inflammation and fibrosis at treatment outset,12 or the presence of extraocular lesions, although this last notion is controversial.6
We have based our therapeutic approach on that described by Foster et al.6,99 Specifically, we use a stepladder approach that takes into consideration severity and progression of the disease, patient’s age, and health, and response to treatment, as follows:
Dapsone is the first-line treatment for patients with early forms of OCP, forms that are slowly progressive, and forms that occur in fragile elderly subjects. The recommended initial oral dose is 100 mg per day, which can be increased in 50-mg increments to 200 mg or even 300 mg. The main adverse effects of dapsone are hemolytic anemia and methemoglobinemia. Dapsone is contraindicated in patients with glucose-6-phosphate dehydrogenase deficiency.
In case of failure or intolerance, dapsone can be replaced by sulfapyridine95 (1 g per day), sulfasalazine95a (3-4 g per day), azathioprine (l-2mg/kg per day), or cyclophosphamide (1-2 mg/kg per day), the latter two drugs being reserved mainly for severe forms (Figs. 4.29 and 4.30). Dapsone can be used as a sparing agent, enabling dose regimen reduction of cytotoxic immunosuppressive drugs. It is very important that patients take the cyclophosphamide tablets in the morning, with abundant beverages in the afternoon to limit the risk of bladder cancer.
In initially severe or rapidly progressive forms the first-line treatment of choice is cyclophosphamide, given orally or as intravenous pulse therapy (1 g per month) (Fig. 4.31). My-cophenolate mofetil therapy also may be useful, but further studies are needed.92a In case of very severe inflammation, systemic corticosteroid therapy can be added (prednisone, 1 mg/kg per day), and progressively tapered. In treatment-resistant OCP, intravenous infusions of pooled human immuno-globulin (2-3 g/kg body weight per cycle, divided over 3 days and repeated every 2-6 weeks) can be a safe and effective therapy.93b
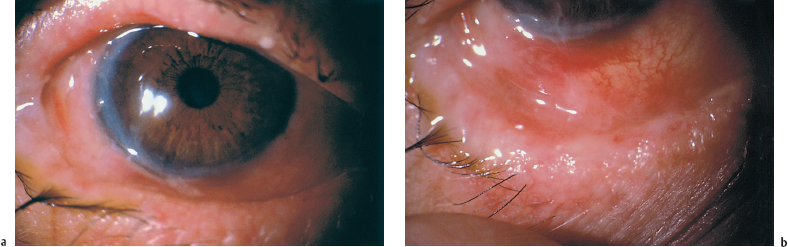
Fig. 4.29a and b Active OCP (despite systemic corticosteroid therapy and dapsone) in a 78-year-old man
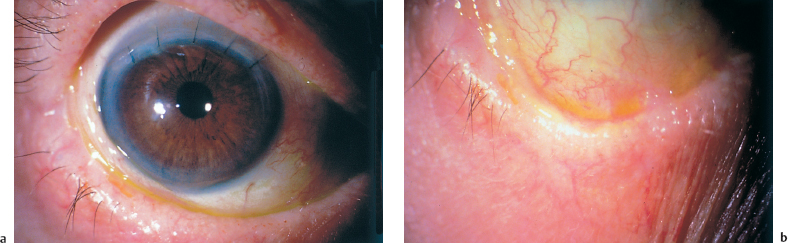
Fig. 4.30a and b Same patient as Fig. 4.29. Cured by adjunction of oral cyclophosphamide (1.5 mg/kg per day). The patient underwent successful cataract surgery
Treatment efficacy is judged on the basis of clinical criteria, including reduction in conjunctival inflammation, prevention of progression of subepithelial conjunctival fibrosis, improvement in ocular symptoms and visual acuity, and reduction in extraocular mucosal lesions. Systemic and biological parameters are used to monitor drug tolerance and adverse effects, and to adjust treatment dosage. Leukocyte and platelet counts should remain above 3500 and 75 000 per mm3, respectively. The immunosuppressive drugs can be withdrawn 3-6 months after all signs of inflammation have vanished and progression of the fibrotic process has been stopped.
Other autoimmune conjunctivitides
Treatment of mild forms of EBA84 is based on dapsone or colchicine. Corticosteroids and/or immunosuppressive drugs are indicated in more severe forms. Systemic cyclosporine is an effective alternative.
Treatment of bullous pemphigoid is based on systemic corticosteroid therapy.84,85,86 Initial treatment with 1 mg/kg per day prednisone is effective in controlling the disease within less than 15 days in two thirds of patients. Although desirable, it is not always possible to discontinue systemic corticosteroid therapy, especially in elderly patients. Other treatments include dapsone, colchicine, erythromycin, tetracycline, and topical steroids. They can be used for localized and mild forms of the disease, or as adjunctive treatments in more severe forms.
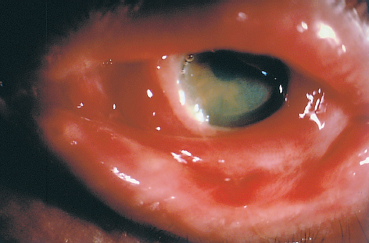
Fig. 4.31 Fulminant OCP: this 75-year-old man progressed from stage I to stage IV in both eyes in less than 6 months. Initial treatment with intravenous cyclophosphamide combined with oral corticosteroids was unsuccessful
Dapsone is the treatment of choice for dermatitis herpeti-formis. A gluten-free diet cures the enteropathy and often leads to a spectacular improvement in mucocutaneous lesions. Noncompliance with this regimen is common and results in relapse of disease.84,85,86
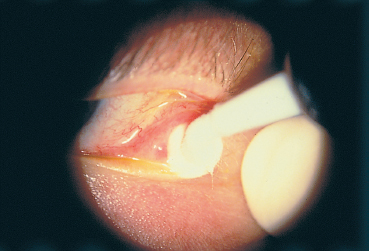
Fig. 4.32 Cryoepilation of ectopic eyelashes
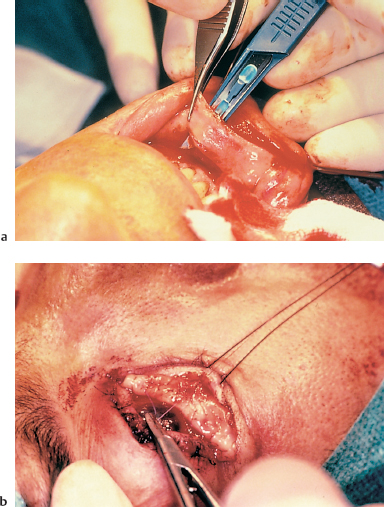
Fig. 4.33a and b Conjunctival fornix reconstruction with a buccal mucosa graft
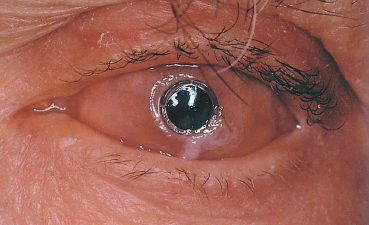
Fig. 4.34 Polytetrafluoroethylene keratoprosthesis for OCP
Systemic treatment of pemphigus vulgaris85,86 is based on corticosteroids (prednisone, 1-2 mg/kg per day), cytotoxic im-munosuppressive drugs, and possibly plasmapheresis. Treatment of the underlying cancer is necessary to cure paraneo-plastic pemphigus. The conjunctivitis in this setting is improved by topical corticosteroid therapy.
Surgical Therapy
Surgery should only be attempted 3-6 months after the inflammatory and fibrotic processes have been controlled, so as to minimize subsequent inflammation. At the time of surgery, some authors add oral corticosteroids to ongoing cytotoxic immunosuppressive therapy.100–103
Palpebral Surgery
In some cases palpebral surgery is required immediately to remove a mechanical irritant which may hinder the assessment of endogenous inflammation.
Several techniques can be used, depending on the lesions:
—Cryoepilation of ectopic eyelashes (Fig. 4.32),
—Marginoplasty, with or without buccal mucosa grafting,
—Eyelash transposition,
—Treatment of a cicatricial entropion,
—Fornix reconstruction, generally with buccal mucosa grafting (Fig. 4.33).
After surgery, immunosuppressive therapy must be continued for 3-6 months.
Ocular Surgery
Superficial keratectomy is indicated for localized corneal pannus; a lamellar or penetrating keratoplasty is performed for more extensive and deeper corneal scars. Limbal stem cell and amniotic membrane grafting are useful adjuncts to restore the ocular surface (see p. 91 ff). In case of too severe corneal lesions such as major neovascularization and xerosis, the only surgical issue is a keratoprosthesis (Fig. 4.34). Recent progress in the biomaterials used for keratoprostheses suggests that human transplants may one day be unnecessary, but follow-up is too short to judge the results of the latest generation of kerato-prostheses.104,105,106
Cataract surgery should also be performed only once the fibrosis has stabilized, in a noninflammed eye. Phacoemulsification with clear corneal incision appears to be the best technique to lower the risk of inflammatory reactivation.106a,106b
The use of an antimetabolite such as mitomycin C or 5-fluorouracil should probably be combined with filtering surgery in patients with glaucoma not responding to medical therapy.
Nonautoimmune Fibrosing Conjunctivitis
Causes
Fibrosing conj unctivitis is not always synonymous with autoimmune conjunctivitis. Many purely ocular or systemic nonautoimmune conditions can induce conjunctival fibrosis, but this latter is generally stable or less progressive, except in some diseases such as Stevens-Johnson syndrome. The main causes of nonautoimmune fibrosing conjunctivitis are listed in Table 4.8. This list is not exhaustive, because all types of chronic conjunctival inflammation theorically can induce fibrosis. Trachoma is the best-known infectious conjunctivitis which is associated with conjunctival scars. Far less well known is the cicatrizing conjunctivitis associated with severe forms of atopy (see Chapter 3) and ocular rosacea (see Chapter 5), the prolonged use of eyedrop preparations (see Chapter 8), and some rare forms of Sjögren’s syndrome. All these conditions can mimick OCP.
Stevens-Johnson syndrome, a fearful condition when severe, is simple to diagnose but raises sufficiently important pathogenic, prognostic, and therapeutic difficulties to merit a separate section within this chapter.
Stevens-Johnson Syndrome
Classification—Pathogenesis
There is a certain confusion surrounding the terminology used to designate this sight- and life-threatening acute bullous mucocutaneous syndrome, which seems either idiopathic or secondary to drug intake or infection. The different names used in the literature are “erythema multiforme,” “Stevens-Johnson syndrome (SJS),” and “toxic epidermal necrolysis (TEN),” also called “Lyell syndrome.” In 1866 Hebra described a self-limited skin disease characterized by papules evolving with a concentric color change or blisters, and a tendency for recurrences.108 Later, in 1922, Stevens and Johnson depicted a more severe febrile illness in two boys, which comprised erosive stomatitis, purulent conjunctivitis, and dissiminated cutaneous eruption.109 Finally, in 1956, Lyell introduced the term “toxic epidermal necrolysis (TEN),” based on observations of four patients with extensive epidermal loss and mucosal involvement.110
According to the last consensus statement111, the classification distinguishes three entities112,113:
—Erythema multiforme minor (von Hebra)
—Erythema multiforme major (Stevens-Johnson syndrome)
—TEN (Lyell)
The definition of erythema multiforme minor only comprises cutaneous involvement which may be isolated or associated, at most, with one mucosal lesion (very rarely affecting the conjunctiva). In contrast, fibrosing conjunctivitis is a common complication of SJS (erythema multiforme major) and TEN, which are more severe forms affecting the skin and several mucosae.114–124 Many authors include erythema multiforme under the term SJS and reserve the term TEN, or Lyell syndrome, for initially very severe forms with extensive bullous cutaneous sloughing (comparable to extensive burns), and in which no mucosal inflammation is seen once the mucosal lesions have healed. Toxic epidermal necrolysis is classically secondary to drug intake, and is believed to involve direct idiosyncratic toxicity. In contrast, the term SJS designates forms with less severe cutaneous involvement, in which the underlying immunological mechanisms would appear to involve circulating immune complexes.125–129 Certain skin lesions would be more characteristic of this syndrome, together with persistent or recurrent inflammation of the mucosae, especially the conjunctiva.130,131 Triggering factors in SJS can be microbial infections, drugs (the same as those incriminated in TEN), some tumors, and some collagen diseases.132–138 A nonexhaustive list is shown in Table 4.9. The main microbial pathogens are Mycoplasmapneumoniae and Herpes simplex.139–144 Between 50% and 60% of cases of SJS are due to a single drug intake, usually involving a sulfonamide, some anticonvulsants (especially hydantoins), barbiturates, and nonsteroidal antiinflammatory drugs (NSAIDs).145–147 Even eyedrops containing compounds such as scopolamine, sulfo-namides, proparacaine, and tropicamide have been incriminated.148–150 However, no trigger is found in many cases. In fact, the distinction between SJS and TEN is academic, and is often difficult and sometimes impossible to make in practice. The underlying mechanism is probably immunoallergic in both cases. This is why it would be better to group together all these mucocutaneous syndromes under a single name such as “Stevens-Johnson-Lyell syndrome” or a more descriptive term not involving proper names.
Table 4.8 Causes of nonautoimmune fibrosing conjunctivitis6
1) Bacterial conjunctivitis 2) Viral conjunctivitis 3) Systemic diseases (see Chapter 9) 4) Allergic-atopic disorders 5) Miscellaneous |
Epidemiology
Stevens-Johnson syndrome and TEN are rare conditions. In the United States the cumulative incidence of SJS and TEN is 4.2 per million person-years. The incidence of TEN is 0.5 per million person-years.146 In France, it is 1.2-1.3 per million person-years.137
These autoimmune bullous skin conditions are more frequent in immunocompromised patients, particularly those with AIDS, with a frequency of 0.95 cases per 1000 patients.135
Stevens-Johnson syndrome is more frequent in males, with a sex ratio of 3 to 1. In contrast, TEN is more frequent in females, with a sex ratio of 1.5 or 2 to 1. The peak of frequency of these syndromes occurs between 20 and 40 years of age,137,146,151-153 but they have also been reported in young children and old people.154 If there seems to be no predilection for a particular race or ethnic group,137,146 a statistically significant association was found between HLA-B12 antigenicity and with SJS and TEN.155,156
Table 4.9 Suspected causative factors associated in Stevens-Johnson syndrome:
1) Infections 2) Drugs 3) Physical Agents 4) Systemic Diseases |
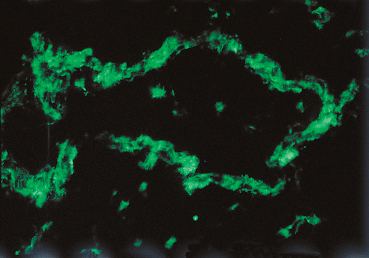
Fig. 4.35 Stevens-Johnson syndrome: immune complex deposits in a conjunctival vessel wall (immunofluorescence)
The incidence of ocular complications varies according to the series, but one can consider that 70-80 % of patients hospitalized for these syndromes have or will have such complications.115–124,151 Contrary to lesions of the other mucosae, oculopalpebral complications do not occur solely during the acute cutaneous phase of the condition, but can also appear some time after the initial episode.130,131,151
Immunopathology
Dermatologists have most extensively studied SJS, an immuno-logically mediated acute bullous mucocutaneous disease. Histological findings in the extraocular mucosae are the same as in the skin. The blister is subepidermal and contains monocytic cells (histiocytes and lymphocytes). There is a perivascular infiltrate composed of mononuclear cells.157,158 During the acute phase of the condition, helper (CD4) T lymphocytes are markedly depleted, with a return to normal counts on clinical recovery. These lymphocytes are abundant in the dermal infiltrate,157–165 where they migrate from the intravascular compartment. It is not known if they are attracted by a primary epidermal modification or if the lymphocytes themselves cause the epidermal lesions. Langerhans cells could also intervene by producing cytokines with chemoattractant properties for lymphocytes.165 Recent studies have shown a rise in suppressor (CD8) T lymphocytes in the exudate contained in cutaneous blisters. These cells also express the surface markers CD45RA and CD29 and are therefore cytotoxic T cells.164
Some authors consider that initial ocular involvement and recurrences are induced by deposition of circulating immune complexes on the vessel walls, provoking “allergic” vasculitis.125,126,129,159 Conjunctival biopsy during the acute phase of SJS shows a nonspecific inflammatory reaction within the conjunctiva, with necrosis of arteries and venules and elastotic degeneration. It has been suggested that the degree of proliferation of epithelial basal cells could correlate with the severity of the condition.151 The chronic phase is associated with sub-epithelial fibrosis and a very marked reduction in goblet cells.6 Immunofluorescence and immunoperoxidase techniques rarely show immune deposits at the BMZ of the conjunctival epithelium, contrary to autoimmune subepithelial bullous conditions. In contrast, immune complexes composed of IgG, IgA, and IgM and complement fractions C3 and C4, have been detected in the vessel walls of the conjunctiva, the mucosae, and the dermis (Fig. 4.35). None of these abnormalities is specific.
Clinical Aspects
The Systemic Disease
Classically, the cutaneous syndrome can occur up to 3 weeks after the first drug intake, whereas it can occur within hours of reexposure to the same drug.123,134 The acute phase lasts an average of 2-4 weeks. For convenience, we will describe SJS and TEN separately, despite the purely theoretical nature of this distinction (Table 4.10).
Table 4.10 Characteristics of Stevens-Johnson syndrome and toxic epidermal necrolysis

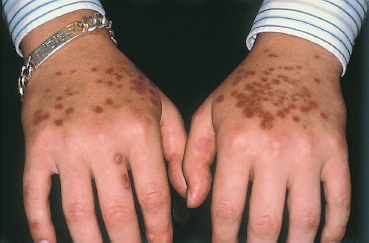
Fig. 4.36 Stevens-Johnson syndrome: cutaneous target lesions
Stevens-Johnson syndrome109,111,115,121
Prodromes generally precede the onset of the skin lesions. This initial phase is often composed of febrile general malaise with headache and sometimes prostration. In typical forms the clinical diagnosis is simple. The skin lesions are usually symmetric and more frequent on the flexor surfaces of the extremities, especially the back of the hands and feet but also the wrists, elbows, and knees. The trunk is only affected in the most severe forms. The eruption starts as a target lesion, is transient, and progresses in certain places to a papular rash (Fig. 4.36). Blisters and bullae can develop, and disappear without scarring. Ungual sequelae are common (Fig. 4.37).
Extraocular mucosal involvement can affect the lips, buccal mucosa (Fig. 4.38), pharynx, larynx, trachea, bronchi, esophagus, and genitalia. They take the form of painful polycy-clic erosions, the vesiculobullous stage being too transient to be visible. They disappear without scarring, contrary to the conjunctival involvement which accounts for the morbidity of the condition, being potentially sight-threatening.
It is during the acute phase of severe forms (erythema multi-forme major) that the vital prognosis is in danger, with a mortality rate as high as 20%. The main complications are pneumonia, septicemia, myocarditis, myositis, and glomerulone-phritis.121
Toxic epidermal necrolysis110–112,120
The general malaise is often more severe than in SJS, combining a burning sensation in the skin and conjunctiva. Initially, the eruption is morbilliform and affects the face and extremities. On coalescing the lesions affect more than 30% of the body surface, forming bullae that detach and leave the underlying dermis naked. The Nikolsky sign is positive. During the acute phase, the most frequently affected mucosa is the buccal mucosa.
Severe, potentially life-threatening infections can complicate the acute phase. Another cause of morbidity is water-electrolyte imbalance.
There are few long-term cutaneous sequelae, the hyper-pigmentation or hypopigmentation disappearing within months. Any scarring is due to superinfections. Some cases of permanent hair loss have been reported.
Ocular Complications
Ocular lesions occurring during the initial phase of the condition must be distinguished from late and chronic complications.
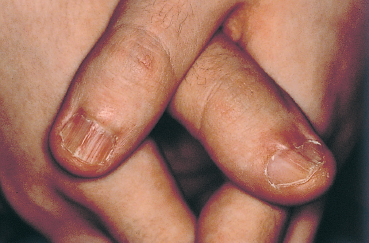
Fig. 4.37 Stevens-Johnson syndrome: secondary nail abnormalities
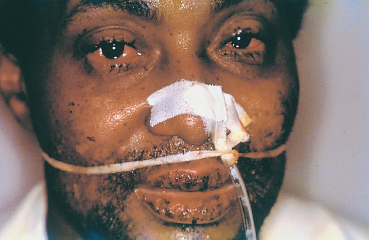
Fig. 4.38 Stevens-Johnson syndrome, acute phase: ocular and buccal involvement
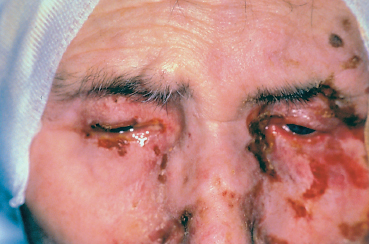
Fig. 4.39 Stevens-Johnson syndrome, acute phase: involvement of the lids, conjunctiva, and skin
Acute ocular complications
The proportion of patients with SJS who develop ocular complications ranges from 15-75%, according to the study.151 Ocular involvement in the acute phase is clinically polymorphous. It can consist of lid inflammation with involvement of the external surface of the eyelids and blepharitis of variable severity (Fig. 4.39). In severe SJS and TEN the most frequent ocular complication is nonspecific conjunctivitis, which sometimes precedes the skin rash. The conjunctivitis can be catarrhal,
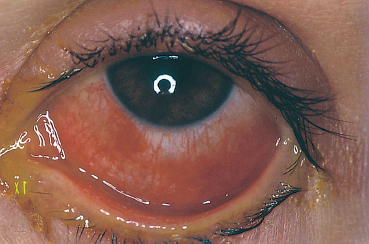
Fig. 4.40 Stevens-Johnson syndrome, acute phase: conjunctivitis
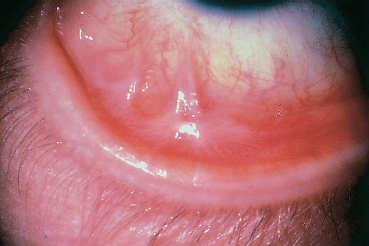
Fig. 4.41 Stevens-Johnson syndrome, inflammatory relapse: fibrosing conjunctivitis with fornix foreshortening and symblepharon formation
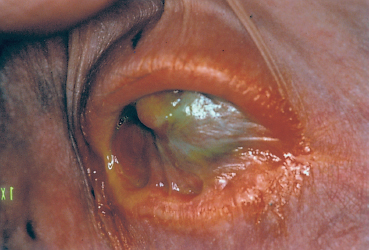
Fig. 4.42 Stevens-Johnson syndrome: ankyloblepharon and xerosis
pseudomembranous, or purulent in the case of bacterial super-infection (Fig. 4.40). Some patients have corneal erosions that persist for 2-4 weeks. Anterior uveitis is rare.151 These complications are generally bilateral. The ocular morbidity of SJS is due to cicatricial conjunctival sequelae.
Chronic ocular complications
Chronic ocular complications of SJS and TEN are potentially severe, often responsible for irreversible loss of vision and blindness. They are characterized by rapidly progressive fibrosing conjunctivitis, with fornix foreshortening, and symblepharon and ankyloblepharon formation151 (Fig. 4.41).
Multiple factors are involved in the loss of corneal transparency:
—Qualitative and quantitative deficiency of the tear film secondary to fibrotic obstruction of the lacrimal gland duct orifices, associated chronic meibomitis, and the loss of goblet cells;
—Conjunctival squamous metaplasia secondary to dryness, and corneal xerosis as final complication (Fig. 4.42);
—Persistent corneal epithelial defects due to mechanical irritation by ectopic lashes, an entropion-trichiasis, or keratinized lid margins;
—Vascularized corneal scars and irregular corneal astigmatism.
Some patients develop recurrent conjunctival inflammation not related to mechanical irritants or to dryness, but perhaps linked to immune complex vasculitis coinciding with recurrent herpes simplex. These ocular recurrences are not associated with cutaneous relapses.130 Cases of late diffuse and occasionally necrotizing scleritis complicating SJS have also been reported.167
Therapy
Medical Therapy
Systemic therapy
Patients are generally hospitalized in intensive care units. Possible culprits must be withdrawn. Any further exposure may induce a relapse of the mucocutaneous syndrome.168–171 Treatment is aimed at compensating for fluid-electrolyte losses due to the epidermal loss. Prevention and treatment of superinfections is essential, as these account for about 50% of deaths during the acute phase.170 Other systemic treatments, especially systemic corticosteroid therapy, are more controversial.172–175 Some studies suggest that high-dose systemic corticosteroids may attenuate epithelial necrosis, but the outcome is not modified by steroid therapy. Moreover, steroids favor superinfections and have other adverse effects too. Other immunosuppressors such as cyclophosphamide176 and cyclos-porine177 have not proved beneficial. The same is true of plasmapheresis.178,179
Ocular therapy
Ocular treatment151 during the acute phase is mainly based on effective lubrication with preservative-free saline, gels, and ointment. Antibiotic eyedrops are only used in case of superin-fection, and cycloplegics are reserved for acute anterior uveitis. Like systemic corticosteroids, topical corticosteroids are controversial in this setting. They can indeed promote conjunctival and corneal superinfections in these patients whose ocular surface epithelium is modified. They have not proved effective in the prevention of symblepharon formation after the acute phase, or during instrumental procedures aimed at preserving the fornices (conformers, mechanical severing of synechiae).151 Topical corticosteroid therapy can at best provide functional relief for patients with severe ocular inflammation. The use of topical cyclosporine has not yet been studied.
Medical therapy of chronic ocular complications focuses mainly on the dry eye syndrome and local superinfections. In this respect it is identical to that indicated during the acute phase of the syndrome. Topical corticosteroids can, in contrast, be used discontinuously to limit the functional discomfort caused by inflammation. Topical all-trans-retinoic acid may attenuate the epithelial transdifferentiation that follows the insult to the conjunctival epithelium, but it is not commercially available.180–183 Topical and systemic mucolytic agents such as N-acetyl-cysteine appear to attenuate mucus strand formation, but their efficacy remains to be proved. In case of persistent severe inflammation or inflammatory relapse resistant to local antiinflammatory drugs, one may use systemic immunosuppression with corticosteroids and/or immunosuppressive cytotoxic agents such as azathioprine and cyclo-phosphamide.176 Treatment guidelines are similar to those of autoimmune bullous disorders.
Surgical Therapy
In the acute phase emergency surgery may consist of lamellar or penetrating keratoplasty for corneal perforation,151 possibly combined with amniotic membrane grafting.184
After the acute phase, when the lesions have stabilized or during the chronic inflammatory phase, reconstructive conjunctivopalpebral surgery and restoration of corneal transparency follow the same rules as those established for autoimmune fibrosing conjunctivitis (see p. 86ff).
Limbal stem cell grafting (homologous rather than auto-logous, because of the more frequent bilaterality of the ocular involvement) can be useful in case of persistent epithelial defects. Patients undergoing this surgical procedure must receive systemic immunosuppressive therapy (oral cyclosporine, 3-5mg/kg per day, or FK-506 [Tacrolimus]) and topical corticosteroids to prevent tissue rejection.185–189 Limbal homograft with a close relative as donor may provide a better outcome.190 Limbal stem cells cultured in vitro,191 amniotic membranes,184,192 and the combination of the two193 have also been used with success in SJS, but published series are small and follow-up is limited.
References
1. Lever WF, Talbott JH. Pemphigus: a historical study. Arch Dermatol Syph 1942; 800-23.
2. Cooper W. Pemphigus of the conjunctivae. Ophthalmol Hosp Rep LondHospRes 1857; 1: 155.
5. Rook A, Waddington E. Pemphigus and pemphigoid. Br J Dermatol 1953; 65: 425-31.
6. Foster CS. Cicatricial pemphigoid. Trans Am Ophthalmol Soc 1986; 84: 527-663.
7. Lever WF. Pemphigus. Medicine 1953; 32: 1-123.
9. Wright P. Cicatrizing conjunctivitis. Trans Ophthalmol Soc UK 1986; 105: 1-17.
10. Ahmed AR, Hombal SM. Cicatricial pemphigoid. Int J Dermatol 1986; 25: 90-6.
12. Mondino BJ, Brown SL Ocular cicatricial pemphigoid. Ophthalmology 1981; 88: 95-100.
44. Gammon WR, Heise ER, Burke WA. Increased frequency of HLA-DR in patients with autoantibodies to epidermolysis bullosa acquisita antigen: evidence that appearance of autoimmunity to type VII collagen is HLAclass II allele associated.J invest Dermatol 1988; 91: 228-32.
84. Fine JD. Management of acquired bullous skin diseases. N Eng J Med 1995; 333: 1475-84.
85. Puissant A, et coll. Dermatologie; ed Ellipses 1987; 45-53.
86. Saurat JH, Grosshans E, Laugier P, Lachapelle JM. Dermatologie-Vénérologie; Masson 1990; 269-72.
88. Hahn JM, Meisler DM, Lowder CY, Tung RC, Camisa C. Cicatrizing conjunctivitis associated with paraneoplastic lichen planus. Am J Ophthalmol 2000; 129: 98-9.
120. Kint A, Geerts ML, de Weert J. Le syndrome de Lyell. Dermatologica 1981; 163: 433-54.
122. Patz A. Ocular involvement in erythema multiforme. Arch Ophthalmol 1950; 43: 244-56.
131. Chan LS, Soong HK, Foster CS. Ocular cicatricial pemphigoid occur-ing as a sequela of Stevens-Johnson syndrome. JAMA 1991; 266: 1543-6.
134. Dunagin WG, Millikan LE. Drug eruptions. Med Clin North Am 1980; 64: 983-1003.
138. Tonnesen MG, Soter NA. Erythema multiforme. J Am Acad Dermatol 1979; 1: 357-64.
139. Shelley WB. Herpes simplex virus as a cause of erythema multiforme. JAMA 1967; 201: 71-4.
144. Rohrer TE, Ahmed AR. Toxic epidermal necrolysis. Int J Dermatol 1991; 30: 457-66.
151. Mondino BJ. Cicatricial pemphigoid and erythema multiforme. Ophthalmology 1990; 97: 939-52.
154. Ginsburg CM. Stevens-Johnson syndrome in children. Pediatr Infect DisJ 1982; 1: 155-8.
185. Thoft RA. Keratoepithelioplasty. Am J Ophthalmol 1984; 97: 1-6.
188. Tsubota K, Satake Y, Kaido M, Shinozaki N, Shimmura S, Bissen-Miyajima H, Shimazaki J. Treatment of severe ocular-surface disorders with corneal epithelial stem-cell transplantation. N Engl J Med 1999; 340: 1697-703.
193. Tsai RJ, Li LM, Chen JK. Reconstruction of damaged corneas by transplantation of autologous limbal epithelial cells. N Engl J Med 2000; 343: 86-93.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


