INTRODUCTION
Extensive regions of the cerebrum, cerebellum, and brainstem participate in controlling gaze. Damage to these areas and their pathway to ocular motor neurons cause disorders of eye motion that are designated as supranuclear when the ocular motor nuclei are spared.
1,2 In this chapter, we describe the clinical effects of lesions in supranuclear ocular motor pathways.
Six eye movement systems are utilized to achieve clear vision; these systems have physiologically and to a large extent anatomically discrete organization in the brain. Systematic examination of each system or class of eye motion is required to localize and diagnose supranuclear disorders. They are the (1) saccadic, (2) smooth pursuit, (3) optokinetic, (4) vestibulo-ocular, (5) vergence, and (6) fixation systems. Four systems generate conjugate movements, called version, while vergence achieves binocular vision by generating disjunctive eye movements that align the two foveas on an object as it approaches or recedes from the head (
Table 10.1). Saccades are fast eye movements that bring the fovea to a target, while pursuit, vestibular, optokinetic, and vergence are smooth eye movements that prevent slippage of retinal images.
1,2 The term gaze refers to the position of the eye in space which is derived from the position of both the eyes in the orbit and the position of the head. Gaze shifts are achieved by head and binocular motion consisting of saccades, smooth pursuit or optokinetic tracking, vergence, and the vesitibuloocular reflex (VOR).
Here we start with the brainstem inputs to the ocular motoneurons that reside in the third, fourth, and sixth cranial nerve nuclei. Then contributions of the cerebellum and cerebral hemispheres are reviewed. At each stage in this bottom-to-top approach, from ocular motoneuron to visual cortex, a summary of the relevant anatomy and physiology is accompanied by descriptions of the effects of discrete lesions and the characteristics of distinctive syndromes.
When the brain initiates any type of eye movement, it must take into account the properties of the eyeball, its suspensory ligaments, fascia and pulleys that guide the tendons of the extraocular muscles.
3 Each muscle has an outer orbital layer that inserts into the muscles’ pulley and an inner global layer that passes through the pulley to insert on the globe. Different populations of ocular motoneurons may supply the global and orbital layers.
4 With the exception of the superior oblique, all muscle pulleys move, which may serve to constrain eye rotations. During fixation, saccades and smooth pursuit movements, eye rotation has only two degrees of freedom, with torsion being constrained according to Listing’s law. Listing’s law states that any eye position can be reached from primary position by rotation of the eye about a single axis lying in the equatorial plane. That is, any position of the eyeball can be reached from primary position by rotation about an axis that is perpendicular to the line of sight in primary position.
5,6 In other words, there is only one fixed torsional eye position for each combination of horizontal and vertical eye position. For the vestibulo-ocular and optokinetic reflexes, however, the torsion constraint of Listing’s law is violated. This physiological violation is determined by the innervation of extraocular muscles in the planes of the semicircular canals. The constraint on torsion is mediated by neural innervation
5,7 in concert with the mechanical properties of the orbital tissue and the action of the pulleys.
3 By using the muscles and pulleys, the brain simplifies its role in controlling three dimensional eye rotations.
Gaze-holding when looking away from the midposition of the orbit demands an appropriate tonic contraction of the extraocular muscles, to prevent the eye from drifting back toward primary position. This gaze-holding function is the achievement of a process called neural integration, where eye velocity signals are transformed into a steady eye position command. The neural integrator function is a distinct property for all eye movements that has its own identified neural substrate in the brainstem.
8,9
EFFECTS OF LESIONS OF THE PONS AND MEDULLA ON HORIZONTAL GAZE
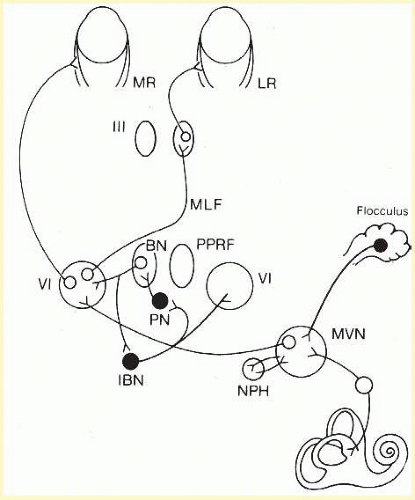
Lesions of the abducens nucleus produce paralysis of both the ipsilateral lateral rectus and contralateral medial rectus for all conjugate eye movements resulting in ipsiversive gaze palsy,
18,19,20,21 but vergence movements are typically spared. Lesions that affect only the abducens nucleus are rare; usually there is also involvement of adjacent structures including the MLF, PPRF, and the facial nerve fascicle.
Lesions of the MLF produce internuclear ophthalmoplegia (INO). The MLF is paramedian in the pons and becomes slightly more lateral in the midbrain as it approaches the oculomotor nuclear complex. INO is characterized by paresis of adduction on the side of the lesion.
2,22 When complete, INO exhibits failure of the adducting eye to cross the orbital midline with saccades, smooth pursuit or vestibular smooth eye movements, and slow adducting saccades up to the midline. The range of adduction in incomplete INO can vary from a few degrees beyond the midposition up to full.
23 The abducting eye, contralateral to an MLF lesion usually exhibits jerk nystagmus (see Chapter 11). This dissociated nystagmus is probably an adaptive phenomenon; as the brain attempts to direct the paretic adducting eye at the visual target, a series of saccades is only evident in the nonparetic, abducting eye.
24 Slow adducting saccades are the minimum diagnostic feature of INO. The term internuclear ophthalmoparesis is used to describe those cases where adduction past the midline is present, but with limitation of amplitude or decreased saccadic velocity. Subtle INO may be detected readily by observing dissociated horizontal optokinetic nystagmus (OKN); the OKN is less intense in the eye on the side of INO when the optokinetic stimulus is moved toward the side of MLF lesion.
25 Repetitive horizontal saccadic refixations can disclose the ocular dysmetria sign of INO
25 consisting of slow hypometric saccades of the adducting eye and concomitant overshoot of the abducting eye. However, even experienced clinicians may miss INO
26 and measurement of saccades to compare adducting and abducting movements is worthwhile.
27,28,29 Magnetic resonance imaging (MRI) with high-resolution views of the brainstem also aids in revealing MLF lesions.
30Skew deviation with hypertropia on the side of the MLF damage sometimes accompanies INO.
31,32 Skew (discussed below) may persist after adduction recovers,
31 and the skew indicates damage to utriculo-ocular static counterroll reflex projections
33,34 that ascend close to the MLF, but are probably outside it, since skew is absent in many cases of INO. Bilateral INO is usually associated with gaze-evoked vertical nystagmus, impaired vertical pursuit, and decreased vertical vestibular-ocular responses
35 because the MLF carries some but not all vertical smooth eye movement signals. Vertical saccades are normal. The vertical vestibulo-ocular reflex (VOR) may show deficient downward eye movements when tested with head impulses.
36 Small-amplitude saccadic intrusions may interrupt fixation.
37 The most frequent cause of INO in young adults, particularly when bilateral, is multiple sclerosis, and in older patients it is infarction (
Table 10.2). Unusual causes compose more than one quarter of cases. The differential diagnosis of INO should be tripartite: multiple sclerosis, stroke, and other causes.
38 Some other causes are listed in
Table 10.2.
Most patients with INO are orthotropic (or exophoric) in primary position without symptomatic diplopia, unless INO is accompanied by skew deviation. Occasionally, patients with bilateral INO show binocular exotropia, designated the WEBINO (wall-eyed bilateral INO) syndrome.
39 The exotropia may also be monocular on the side of a unilateral MLF lesion, and designated as the WEMINO syndrome.
40 The explanation for the exotropia is unclear because monkeys with a lidocaine-induced internuclear ophthalmoplegia show an
increased accommodative vergence to accommodation ratio, implying that the MLF actually carries signals that inhibit vergence.
22 Thus, the cause of exotropia in INO is uncertain and we have observed it in patients with preserved convergence. Furthermore, the former classification of INO into anterior and posterior types depending on the integrity of the convergence mechanism is not of localizing value in identifying the rostral-caudal location of the MLF lesion. The so-called posterior internuclear ophthalmoplegia of Lutz
41 in which abduction (but not adduction) is impaired is rare. It has been difficult to conceptualize because the abducens contains neurons that innervate both lateral and medial rectus muscles for all conjugate eye movements.
42 One possible explanation concerns oculomotor internuclear neurons, which project from the third nerve nucleus down to the contralateral abducens nucleus; inactivation of these internuclear neurons causes impaired abduction.
43A combined lesion of one MLF and the adjacent abducens nucleus or PPRF produces paralysis of all conjugate movements except for abduction of the eye contralateral to the side of the lesion—the
one-and-a-half syndrome.
44,45,46 Marked exotropia of the eye on the side opposite that of the brainstem lesion is a feature termed
paralytic pontine exotropia.
47 This occurs with brainstem infarction, hemorrhage, multiple sclerosis, cavernoma, and glioma; rarer causes are basilar artery aneurysm, and tumor.
Discrete lesions of the PPRF
48,49—which mainly corresponds to the nucleus pontis centralis caudalis and contains saccadic burst neurons (
Fig. 10.2)
50—cause loss of saccades and quick phases of nystagmus to the side of the lesion. Vertical saccades are misdirected obliquely away from the side of the lesion.
51 Selective slowing of horizontal saccades occurs in degenerative conditions, such as some variants of spinocerebellar atrophy.
52,53,54,55 Causes of slow or absent saccades are summarized in
Table 10.3. Infarction of the paramedian pons usually, but not always, also involves adjacent fibers conveying vestibular and pursuit inputs to the abducens nucleus so that ipsiversive gaze palsy includes saccades, smooth pursuit and the VOR
51,56,57,58 (
Table 10.4). Pontine disease may cause an ipsiversive defect of smooth pursuit by affecting the dorsolateral pontine nuclei and their projections to the cerebellum (see below). More rostral brainstem lesions may cause ipsiversive smooth pursuit deficits, whereas caudal brainstem lesions tend to cause contraversive deficits
57,59; this is discussed further below.
Hereditary dysgenesis of brainstem pathways can cause familial horizontal gaze palsy.
60 Saccades, smooth pursuit, and the VOR are paralyzed in both directions, but vertical gaze is intact. Progressive scoliosis and low amplitude horizontal pendular nystagmus are also features of this autosomal recessive genetic disorder. It results from mutations in the ROBO3 gene on chromosome 11q23-25 required for hindbrain axon midline crossing.
61,62,63 Hypoplasia of the pons and medulla with a deep midline pontine cleft (split pons sign) and butterfly configuration of the medulla are evident on MRI.
64 Developmental failure of crossing of ocular motor pathways in the brainstem is the mechanism of the horizontal palsy. Only vergence eye motion is spared, and it may substitute for the loss of horizontal version,
60 as discussed later. We have also recognized a familial paralysis of vertical gaze associated with dysphonia, dysarthria, and limb spasticity.
65 Inheritance is autosomal dominant, and it is not related to the genetic lipidoses, but the molecular basis of this syndrome is not known. Gaze palsy may be a sign of lipid storage diseases.
66 Vertical gaze paresis with foam cells or sea-blue histiocytes in the bone marrow is a neurovisceral storage disease with profiles of lipid analysis similar to Niemann-Pick disease, type C (NPC).
67 Paresis of horizontal saccades is a feature of Gaucher’s disease (GD).
68 Saccade initiation failure is often the earliest neurological sign in GD type 3.
Gaze evoked nystagmus is a sign of damage to the lateral pontomedullary junction and its cerebellar connections. Bilateral experimental lesions of the nucleus prepositus hypoglossi and adjacent medial vestibular nucleus (
Fig. 10.1) abolish the gaze-holding mechanism of the neural integrator for horizontal eye motion.
8,69 The neural integrator transforms an eye velocity signal into the eye position command necessary to hold the eye steady in an eccentric orbital position. After damage to the neural integrator network (which includes connections with the vestibular cerebellum), the eye cannot be held in an eccentric position in the orbit and it drifts back to primary position. Corrective quick phases then produce gaze-evoked nystagmus (see Chapter 11) as the integrator leaks away its tonic eye position command. Disease affecting the vestibular nuclei and nucleus prepositus hypoglossi cause vestibular imbalance—manifest as nystagmus or skew deviation, and impairment of gaze-holding. This occurs with lateral medullary infarction (Wallenberg’s syndrome) in which the spontaneous nystagmus is usually horizontal or mixed horizontal-torsional with the slow phases directed toward the side of the lesion
70,71; the nystagmus may reverse direction in eccentric positions, suggesting coexistent involvement of the gaze-holding mechanism. Skew deviation, with an ipsilateral hypotropia, cyclodeviation (lower eye is more extorted), and ipsilateral head tilt (when present together, called the ocular tilt reaction [OTR]) reflect an imbalance of otolithic inputs.
33,34,72 In addition, patients with Wallenberg’s syndrome show a characteristic ipsipulsion, the common type of lateropulsion in which the eyes deviate conjugately toward the side of the lesion during vertical or horizontal saccades, or eyelid closure.
70,73,74,75 A possible explanation for ipsipulsion is that infarction of the inferior cerebellar peduncle leads to increased inhibition of the fastigial nucleus by the cerebellar vermis.
73,76,77 This saccadic dysmetria is discussed further below in the section dealing with cerebellar influences on eye movements.
VESTIBULO-OCULAR REFLEX DISORDERS
Head rotation elicits the angular vestibulo-ocular reflex disorders (VOR). The ratio of the output of the angular VOR (smooth eye movement speed in one direction) to the input of the reflex (head speed in the opposite direction) is its gain. VOR gain must approximate 1.0 in order to prevent slippage of retinal images and maintain clear vision. The eyes and head must also be 180 degrees out of phase; this normal phase difference is called zero, by convention. Abnormal gain or phase of the reflex causes visual blur and oscillopsia. Nystagmus quick phases correct for failures of smooth eye movement motion to match head speeds. Imbalance of the VOR is caused by damage to the vestibular labyrinthine or nerve on one side or by asymmetric damage to its central brainstem connections to the ocular motor nuclei. This imbalance creates jerk nystagmus beating toward the side of defective vestibular smooth eye motion (see Chapter 11). Here we outline tests of the VOR that are important in assessing supranuclear impairment after brainstem lesions.
Low gain of the VOR signifies bilateral peripheral vestibular or brainstem disease. Acute unilateral labyrinthine or vestibular nerve damage lowers gain acutely, but compensation occurs within days or weeks so that a gain asymmetry of the reflex persists only during high frequency (> approximately 2 Hz) head motion. High gain of the VOR (>1.0) is uncommon and is a feature of cerebellar disease.
78The oculocephalic reflex (OCR) elicited by passive head on body rotation (doll’s head reflex) tests the range of the VOR. Binocular limitation of the range of the VOR indicates supranuclear disruption of brainstem VOR pathways. Conversely normal range of the OCR, in the presence of limited range of saccades or smooth pursuit confirms that a lesion responsible for the gaze paresis is supranuclear. However the OCR does not assess the VOR alone. Passive movement of the head at low frequencies activates visual following responses, both smooth pursuit and optokinetic, as well as the VOR. If the VOR is absent, visual following reflexes can move the eyes at the same velocity as the head if the motion is below 1 Hz. Only in comatose patients do low frequency passive OCR movements test the VOR alone.
Visual acuity during head shaking also assesses the VOR. When the patient is instructed to shake their head from side to side at 2 to 3 Hz through an amplitude of about 20°, Snellen visual acuity should remain about the same as with the head immobile. If acuity falls by two or three lines the VOR gain is low or high, or its phase is abnormal. If the VOR is hyperactive (gain > 1.0) the patient perceives movement of the image in the same direction as head movement. If the VOR is hypoactive (gain < 1.0) images appear to move opposite to the direction of head motion. One can also examine the eyes for nystagmus. If the VOR is normal while the patient fixates an object during high frequency head oscillation, the eyes should remain stable in space. Vestibular nystagmus indicates abnormal VOR gain or phase.
Ophthalmoscopy during Head Shaking is another technique for detecting VOR abnormalities. This involves examination of one fundus while the patient shakes his head from side to side at 2 to 3 Hz. If the VOR is normal the optic disc (or other fundus landmark) remains stable.
79 If the VOR is overactive (gain > 1.0) the disc moves in the same direction as the head whereas if the VOR is hypoactive (gain < 1.0) the disc moves opposite to the direction of head movement. Care must be taken that the head does not sway from side to side, since head translation causes the disc to move, even when the VOR is normal. This test can be difficult to perform and interpret. Patients must wear their regular glasses during the test, since the gain of the VOR is adapted according to the magnifying power of their lenses.
Post head shaking nystagmus also occurs after brainstem or cerebellar damage. Patients with unilateral peripheral vestibular lesions have nystagmus directed away from the affected side after vigorous head shaking.
80 The patient is instructed to shake his head through 40 degrees of range at about 2 Hz for 20 seconds. Then when the head is stopped, the observer examines the patient’s eyes under Frenzel glasses (plus 20 lenses which magnify the eyes and blur the patient’s fixation). Slow phases of head shaking nystagmus (HSN) are directed toward the impaired ear. It lasts less than 20 seconds, and is sometimes followed by a low amplitude reversed HSN with slow phases away from the impaired ear. After central brainstem or cerebellar lesions HSN may be directed in planes different from the direction of head shake, or with slow phases away from the side of brainstem damage.
81,82,83The head impulse test usually assesses a severe unilateral peripheral vestibular loss but can be abnormal after central damage. While the patient fixates a distant target the head is briskly moved to one side. Normally the eyes should stay on target and the examiner should see only a smooth compensatory eye movement. Patients with unilateral vestibular loss make one or more saccades to bring the eyes on target when the head is moved toward the side of the impaired ear. Saccades in the same direction as the VOR signify inability of vestibular smooth eye movements to match head velocity.
84,85 Thus a patient with right vestibular neuritis will make leftward saccades during rightward head movements. Compensatory, refixation saccades after rapid horizontal head rotations indicate a horizontal semicircular canal or vestibular nerve lesion on the side toward which the head is being rotated. The head must be moved at high velocity, because the VOR eventually becomes rebalanced for low velocity head movements after unilateral peripheral vestibular damage. An abnormal test may also indicate brainstem or cerebellar disease.
86,87
BRAINSTEM CONNECTIONS FOR VERTICAL AND TORSIONAL MOVEMENTS
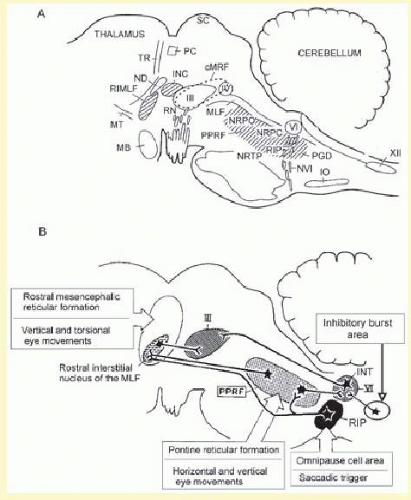
Vertical and torsional saccadic commands and the vertical gaze-holding signal are synthesized in the midbrain, while vestibular and pursuit signals ascend to the midbrain from the lower brainstem. The ocular motoneurons that control vertical and torsional eye movements lie in the oculomotor and trochlear nuclei. Vertical and torsional saccade commands to them are generated from the
rostral interstitial nucleus of the medial longitudinal fasciculus (riMLF) which lies within rostral extent of the MLF, rostral to the tractus retroflexus and caudal to the mammillothalamic tract (
Fig. 10.2).
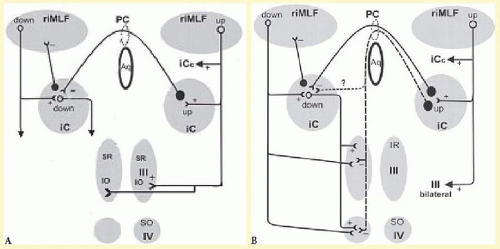
88,89 The riMLF contains excitatory burst neurons for vertical and torsional saccades and nystagmus quick phases. Each riMLF contains neurons that burst for upward and downward eye movements but for torsional quick phases in only one direction. Thus, the right riMLF discharges for quick phases that are directed clockwise with respect to the subject; that is with the upper poles of the eyes rotating toward the right shoulder.
90 In addition, each riMLF is connected to its counterpart by the posterior commissure (
Fig. 10.3), and by a more rostrally located ventral commissure that lies ventral to the aqueduct. The riMLF projects to motoneurons innervating elevator muscles bilaterally but to motoneurons innervating depressor muscles ipsilaterally.
91,92 Furthermore, each burst neuron in the riMLF appears to send axon collaterals to motoneurons supplying yoke muscle pairs (e.g., superior rectus and inferior oblique); this is the neural substrate whereby Hering’s law for conjugate motion applies to vertical saccades.
The riMLF also projects to the
interstitial nucleus of Cajal (INC), which lies just caudal to it (
Fig. 10.2). The INC has an important role in integrating eye velocity commands into vertical gaze-holding (as the neural integrator).
93,94 In addition, the INC also receives inputs that ascend from the vestibular nuclei.
9 The INC projects to motoneurons of the vertical ocular motor subnuclei. The projections of the INC to the elevator ocular motor nuclei (superior rectus and inferior oblique) pass dorsally over the aqueduct in the posterior commissure, (
Fig. 10.3) and this might explain why lesions of the posterior commissure limit mainly upward eye movements.
93,95,96,97 The INC also contains neurons that project to motoneurons of the neck and trunk muscles and appears to coordinate combined torsional-vertical movements of the eyes and head.
98 The neural signals necessary for
vertical vestibular and smooth pursuit eye movements ascend from the medulla and pons to the midbrain. The MLF is the most important route for these projections but the brachium conjunctivum and other pathways are also involved.
14
Effects of Brainstem Lesions on Vertical Gaze and Ocular Torsion
Unilateral, experimental
lesions of the riMLF cause a mild defect in vertical saccades, because each nucleus contains burst neurons for both upward and downward movements. On the other hand, unilateral riMLF lesions produce a specific defect of torsional quick phases.
90 For example, with a lesion of the right riMLF, torsional quick phases, clockwise from the point of view of the subject (extorsion of the right eye and intorsion of the left eye) are lost and contralesional beating torsional nystagmus may be evident.
99 In contrast lesions of the INC are associated with ipsilesional torsional nystagmus. Either riMLF or INC lesions on one side cause contralesional tonic torsional deviation of the eyes.
99 Vertical saccadic palsies in patients with unilateral lesions of the riMLF or its immediate connections in humans probably reflect involvement of the commissural pathways of the riMLF that makes the lesion, in effect, bilateral.
100 In general, bilateral lesions are required to produce clinically apparent deficits of vertical eye movements.
101 Bilateral experimental lesions in the region of the riMLF in monkeys cause a vertical saccadic deficit, that may be more pronounced for downward eye movements
102; this may be because upward saccadic projections are bilateral whereas downward saccadic projections are unilateral (
Fig. 10.3).
91,92,103 With bilateral riMLF lesions, vertical gaze-holding, vestibular eye movements, and possibly pursuit are preserved, as are horizontal saccades, with the deficits limited to either downward or both upward and downward saccades.
103,104 Certain metabolic and degenerative disorders may lead to selective slowing or absence of vertical saccades (
Table 10.3).
Unilateral experimental lesions of the INC are reported to impair gaze-holding function in the vertical plane, reflecting its role as a key element of the velocityto-position integrator for vertical gaze, in concert with the vestibular nuclei.
94,105 Another cell group important for neural integration is in the paramedian tracts which lie within the MLF and project to the flocculus.
106,107 Chemical lesions of these tract neurons in cats lead to vertical gaze evoked nystagmus and primary position downbeat nystagmus.
108 Bilateral
lesions of the medial longitudinal fasciculus (causing bilateral INO) in patients impair vertical vestibular smooth eye movements and smooth pursuit but spare vertical saccades.
35,109 In addition, vertical gaze-evoked nystagmus occurs in bilateral INO as the result of partial loss of the vertical eye position signal caused by disrupting projections from the vestibular nuclei to the INC or by damaging the paramedian tracts in the MLF.
Lesions of the
posterior commissure cause loss of upward gaze
95,97 usually all types of eye movement are affected, although the upward range of the VOR and smooth pursuit and Bell’s phenomenon may be spared. Experimental inactivation of the posterior commissure with lidocaine impairs vertical gaze-holding (neural integrator) function.
96 Typical findings with posterior commissure damage constitute the pretectal syndrome described next.
Pretectal Syndrome
Lesions dorsal to the aqueduct of Sylvius in the rostral midbrain produce a distinctive combination of neuroophthalmologic signs involving upward gaze, eyelids, pupils, accommodation, and vergence.
97,110,111 This pretectal syndrome (
Table 10.5) encompasses many eponymic and anatomical designations, such as Parinaud, Koerber-Salus-Elschnig, Sylvian aqueduct, dorsal midbrain and posterior commissure syndromes.
39 Pineal area tumors and midbrain infarction
103,104 are the most common causes.
110,111,112 Other etiologies include congenital aqueductal stenosis, multiple sclerosis, syphilis, arteriovenous malformations, midbrain hemorrhage,
113 encephalitis, midbrain or third ventricle tumors, herniation of the uncus,
114 and lesions resulting from stereotactic surgery.
115 The pupils are mid-position or large and demonstrate light-near dissociation; that is, the light reaction is smaller than the near response. Pathologic lid retraction (Collier’s sign; the posterior fossa stare) and lid lag are common findings (
Table 10.5). Paralysis of upward gaze is the hallmark sign. Upward saccades are lost above the orbital midline, whereas the range of upward pursuit and the visually enhanced VOR (V-VOR) as assessed during oculocephalic testing in the light are often spared. Eye movement recordings, however, reveal lowered speed and phase lead of the VOR.
116 Both smooth pursuit and vestibulo-ocular responses and Bell’s phenomenon, may become paralyzed above the midline. Attempts at upward saccades evoke bursts of convergence-retraction oscillation (nystagmus). The oscillations are readily elicited with down-going optokinetic targets providing a stimulus for repetitive upward saccades; each fast phase is replaced by a convergence or retraction movement. There is debate as to whether it is a true form of nystagmus,
117 or a series of opposed adducting saccades, followed by slow divergence movements
118 (see Chapter 11). This is in contrast to normal subjects who show transient convergence during downward saccades and divergence during upward saccades.
119 Rarely, a divergence-retraction nystagmus may occur in patients with the pretectal syndrome. Cerebral hemispheric lesions associated with focal motor seizures may cause retraction nystagmus temporally associated with periodic lateralized epileptiform discharges (PLEDs) on the electroencephalogram.
120Unilateral lesions in the midbrain can rarely result in a monocular supranuclear defect of supraduction called “
double elevator palsy,” consisting of weakness in both elevators on the affected side—the superior rectus and the inferior oblique muscles. The lesion is supranuclear in the rostral mesencephalon in the pretectum,
121 and not nuclear, because one oculomotor nuclei innervates the inferior oblique muscle on the same side and the contralateral superior rectus muscle and one would expect crossed elevator deficits in partial unilateral oculomotor nuclear lesions. Also, in the primary position, the eyes are aligned straight without any vertical strabismus and Bell’s phenomenon is preserved, which support supranuclear paresis.
121,122,123 The pathophysiology of supranuclear double elevator palsy is not clearly understood. It was hypothesized that efferent fibers from the riMLF destined to the homolateral superior rectus and contralateral inferior oblique subnuclei may be interrupted, to explain the contralesional double elevator palsy.
123 This is supported by other single case reports, where the lesion was contralateral to monocular elevation paresis.
124,125 Double elevator palsy associated with contralateral Horner’s syndrome and abduction deficits also localizes to a contralateral lesion.
125 However, monocular elevation palsy is also reported with an ipsilateral midbrain lesion, and damage to the upgaze efferent fibers from the riMLF has been postulated.
126 The mechanism of this monocular supranuclear entity is debated.
Subaqueductal Syndrome
Lesions in the midbrain tegmentum, ventral to the aqueduct of Sylvius and rostral to the third nerve nucleus, can cause selective paralysis of downward saccades by disrupting projections from the riMLF to the INC and oculomotor and trochlear nuclei.
102,104,127 The riMLF itself can be spared when downward gaze is paralyzed.
128 Since one riMLF projects to motoneurons innervating elevator muscles bilaterally but to motoneurons innervating depressor muscles ipsilaterally, downward saccades are more vulnerable to lesions affecting it’s projections.
91,92,103,129 More often both upward and downward saccades and pursuit, and convergence are paralyzed. The pupils are small or mid-sized and react poorly to both light and near stimuli (
Table 10.5).
The responsible lesions are usually bilateral,
104 but unilateral paramedian infarcts of the ventral midbrain can also paralyze downward gaze or both upward and downward gaze probably by interrupting commissural connections to nuclei on the other side.
100,130,131 Infarction in the distribution of the thalamo-subthalamic artery of Percheron is usually responsible for the subaqueductal syndrome.
100,132,133,134 Patients are often in stupor or a state of akinetic mutism; a selective down gaze palsy may be the critical localizing sign, although it is sometimes overlooked in intensive care units. In contrast, patients with the pretectal syndrome are usually alert. In the subaqueductal syndrome, dissociated palsy of vertical saccades consisting of loss of voluntary and visually guided upward and downward saccades with preserved quick phases of the vertical and torsional VOR
134 can result from disruption of descending corticofugal saccadic pathways in the rostral midbrain to the nucleus reticularis tegmenti pontis (NRTP) and riMLF (
Fig. 10.2). Preservation of the riMLF itself and of ascending projections from the PPRF to the riMLF are indicated by the preserved VOR quick phases.
134 This site of lesion can also paralyze upward and downward smooth pursuit and catch-up saccades while sparing the range and speed of vertical and torsional vestibular reflex smooth eye movements.
134In both the pretectal and subaqueductal syndromes (
Table 10.5), the vertical VOR or smooth pursuit may appear to be spared.
112 However, reduced gain (the ratio of eye velocity to head velocity), limited amplitude and abnormal phase lead of the vertical VOR occur with rostral midbrain lesions that spare the oculomotor and trochlear nuclei.
100,112,116 This indicates that supranuclear lesions of the rostral midbrain can impair the vertical VOR and contradicts the traditional concept that supranuclear lesions spare the vertical VOR (although it sometimes does spare its range and speed
134). This misconception results from the usual method of testing the vertical VOR at the bedside: the oculocephalic maneuver, in which the patient’s head is passively flexed and extended at a low speed while fixating on a target in a well-lit room. As noted above, the eye movements that result are generated not only by the vertical VOR, but also by visual enhancement of this reflex by smooth pursuit and optokinetic or ocular following system smooth eye movements.
135,136 At low rates of head rotation (below 1 Hz), V-VOR can operate with a full range of ocular excursion when the VOR is severely reduced. Thus, although the examiner may judge the amplitude of oculocephalic eye movements to be normal, the gain and phase of the vertical VOR may be abnormal but not detected at the bedside.
112,116Midbrain lesions ventral to the aqueduct are often associated with nuclear or fascicular oculomotor or trochlear nerve palsy, but here we have confined our discussion to lesions rostral to the oculomotor nucleus, as causing genuine supranuclear vertical gaze defects of the pretectal and subaqueductal syndromes. Associated supranuclear paresis of horizontal gaze occur with lesion in the rostral midbrain reticular formation.
137,138 The saccadic paresis is contraversive, and pursuit paresis may be acutely contraversive, then bidirectional or ipsiversive. Cerebral cortiocofugal projections to the NRTP or projections from the superior colliculus to the PPRF may be responsible for midbrain paresis of horizontal saccades. Clinically the horizontal V-VOR is typically full in range, but lesions in the rostral midbrain reticular formation of monkeys cause loss of the contraversive VOR.
139
Sustained Vertical Deviations
A number of disparate conditions may lead to tonic upward or downward eye deviations. One dramatic example is the sustained upward deviation of oculogyric crisis (see below under
Parkinson’s Disease and Parkinsonism). Otherwise, tonic up-gaze deviation is seen in unconscious patients, especially after hypoxic-ischemic brain injury, perhaps reflecting loss of cerebellar Purkinje cells that normally balance vestibular and gaze-holding mechanisms. This notion is supported by the observation that patients who survive such insults develop downbeating nystagmus.
140 Tonic downward deviation may occur transiently in neonates and does not necessarily indicate a neurologic abnormality; in such cases, the eyes can be easily driven above the horizontal meridian by the vertical doll’s-head maneuver.
141 In comatose patients, sustained downward deviations, with small unreactive pupils, usually accompany bilateral thalamic infarction or hemorrhage.
142 The setting sun sign of the eyes in infantile hydrocephalus is a clinical phenomenon due to pathologic eyelid retraction and upward gaze palsy. Reversibility of this sign with ventricular decompression indicates a dynamic mechanism secondary to acutely increased intracranial pressure that distends the third ventricle and compresses the posterior commissure.
143 Tonic downward deviation may occur with metabolic encephalopathies.
144 These sustained downward deviations are to be distinguished from “ocular bobbing,” an oscillatory disorder consisting of an abrupt downward jerk followed by a slow upward drift to midposition (see Chapter 11).
Skew Deviation and the Ocular Tilt Reaction
Skew deviation is defined as vertical strabismus caused by supranuclear lesions. Damage to vestibulo-ocular pathways between the labyrinth and neurons of the ocular motor nuclei is responsible. Lesions involve the brainstem and its cerebellar connections. The misalignment may be the same in all positions of gaze (comitant), may vary with gaze position, or may even alternate, showing right hypertropia on rightward gaze and left hypertropia on leftward gaze.
145,146,147 Alternating skew is typically a sign of pretectal, cerebellar or cervicomedullary junction pathology. Skew must be distinguished from trochlear nerve palsy and from peripheral neuromuscular disorders such as thyroid ophthalmopathy and myasthenia gravis. Indeed, the diagnosis of skew deviation is contingent upon exclusion of involvement of peripheral nerves, neuromuscular junction, and extraocular muscles as the cause of vertical misalignment of the visual axes.
Skew deviation is an element of the OTR when it is accompanied by head tilt and ocular torsion
32,34,148 (
Fig. 10.4) with all three elements being originally attributed to a central imbalance of input from the utricle of the inner ear to the oculomotor and trochlear nuclei.
148 Both eyes rotate in same direction as the head is tilted and toward the lower skewed eye. Although skew deviation is accompanied by abnormal torsion of both eyes in the same direction, the torsion is often unequal in the two eyes (dysconjugate), and head tilt is usually absent. Tilt of the subjective perception of the visual vertical away from the side of hypertropic eye is also a feature of skew deviation, either alone or accompanied by head tilt in the OTR.
32,149 Experimental stimulation of one utricular nerve in the cat produces elevation of the ipsilateral eye (hypertropia), depression of the contralateral eye (hypotropia), and torsion of both eyes with their upper poles rolling contralaterally.
150 Unilateral stimulation in the midbrain tegmentum, near the INC, causes an OTR with ipsilateral hypotropia and contralateral hypertropia while the head tilts and the eyes roll toward the side of stimulation.
151,152 These observations indicate a decussation of excitatory utriculo-ocular reflex projection, and evidence from lesions in patients indicates that projections from one utricle to the ipsilateral vestibular nuclei later decussate in the tegmentum of the pons while ascending to the contralateral oculomotor and trochlear nuclei.
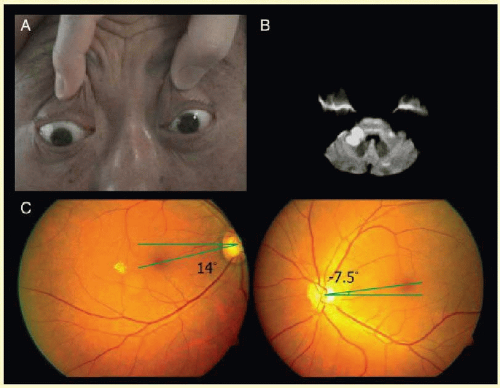
153Distinction of skew deviation from fourth nerve palsy is made by the three step test for strabismus and by the observation of intorsion of the higher eye in skew but extorsion of the higher eye in fourth nerve palsy. This difference in torsion may be detected by funduscopy and fundus photography
154 (
Fig. 10.5); by visual field assessment
155 showing elevation of the blind spot with skew deviation and depression of the blind spot with fourth nerve palsy; and by the use of double Maddox rod technique. If a head tilt is present it is away from the side of the higher eye in both fourth nerve palsy and skew deviation. In fourth nerve palsy the head tilt is compensatory to monocular excyclotorsion of the hypertropic eye and in the opposite direction, but in skew deviation the head tilt is in the same direction as binocular torsion; both the head tilt and torsion arise from an imbalanced static utriculo-ocular counterroll reflex.
33,34 A report
156 of a postural test to distinguish skew deviation from vertical strabismus caused by fourth nerve palsies indicated that when patients with skew deviation are moved from the head upright to the supine position their vertical misalignment and ocular torsion decrease or stop. In contrast the vertical misalignment and ocular torsion in fourth (trochlear) nerve palsy remains the same in the erect and supine positions.
156 It has been proposed that a vertical deviation that decreases by over 50% from the upright to supine position indicates skew deviation.
157 Perhaps changing from an upright to supine position, by moving the orientation of the utricles from earth-horizontal to earth-vertical, reduces asymmetry of the utriculo-ocular reflex causing reduction vertical misalignment and torsion with skew deviation but not with fourth nerve palsy.
157 On the other hand, the supine position might not alter tonic imbalance of input from the utricular nerves to ocular motoneurons caused by central (brainstem or cerebellar) lesions, and our experience is that skew deviation remains the same in the erect and supine positions.
The OTR is typically sustained,
33,153,158,159 but it can be paroxysmal.
148,160 Occasionally skew deviation slowly alternates or varies in magnitude over the course of minutes.
161,162,163,164 The periodicity of the phenomena is reminiscent of periodic alternating nystagmus (see Chapter 11) and the two phenomena may coexist.
165 In cases when the skew deviation is paroxysmal, the mechanism is thought to be irritative.
148,160 This interpretation is supported by observations that stimulation in the region of INC caused an (OTR), with episodes of contralateral hypertropia and ipsilateral head tilt.
151,152Brainstem lesions cause most cases of skew deviation,
33,153,166 but focal cerebellar
167,168 or unilateral peripheral vestibular damage
169 are sometimes responsible. Considerable evidence
33,170 has accrued to confirm the postulate
148,166 that disruption of the static ocular counterroll reflex mediated through the utriculo-ocular pathway
171 is responsible for the OTR
148 and for skew deviation without head tilt.
166 Although focal cerebellar damage alone can be responsible,
167 the specific cerebellar regions involved remain to be established.
33,167,168,172 Infarction of the nodulus is associated with contralesional tilt of the subjective vertical and skew deviation.
173 In patients with acute vertigo, nystagmus, nausea or vomiting, head-motion intolerance, and unsteady gait, skew deviation can identify brainstem stroke when an abnormal horizontal head impulse test falsely suggests a peripheral vestibular disorder. In this acute vestibular syndrome a three-step bedside ocular motor examination called HINTS (Head-Impulse-Nystagmus-Test of Skew) is reported to rival the sensitivity of early MRI in detecting brainstem stroke.
174 Nonetheless clinicians should be mindful that acute unilateral labyrinthine damage can also cause skew deviation, with ipsilateral hypotropia and ipsiversive ocular torsion.
169