Chapter 35 Stem Cells and Cellular Therapy
 For additional online content visit http://www.expertconsult.com
For additional online content visit http://www.expertconsult.com
Stem cells as therapeutics to treat retinal disease
Nothing more dramatically captures the imagination of visually impaired patients or the ophthalmologist treating them than the possibility of rebuilding a damaged retina with “stem cells.” Since many retinal neuro- and vasculodegenerative diseases progress slowly, it may be possible to use stem cell-derived “replacement cells” to prevent visual loss if such therapies are performed at an early stage of disease. Defined as pluripotent, self-renewing cells capable of differentiating into a variety of cell types, stem cells can be derived from early embryos and, under appropriate conditions, will differentiate into a variety of tissues, including mesoderm (e.g., muscle), endoderm (e.g., the gastrointestinal tract), ectoderm (e.g., skin) and neuroectoderm (e.g., brain and retina). Stem cells have also been identified and isolated from adult tissues and presumably represent a pool of progenitor cells that may serve to maintain a supply of cells to maintain various tissue types as well as rescue/repair damaged tissue following injury or stress. In addition to “developmentally arrested” adult stem cells that reside in normal adult tissues, a population of pluripotent stem cells can now be derived from adult somatic tissues. These stem cells, called induced pluripotent stem cells (iPSC) represent a highly significant advance in our ability to derived autologous replacement tissues for the treatment of a variety of ocular, and other, diseases and will be discussed in greater detail below. Overall, there are now several populations of stem cells that have been described and each may have relative benefits for the treatment of different diseases1. Readers are referred to Chapter 125 (Transplantation Frontiers) for further discussion of clinical applications of retinal progenitor and adult transplantation therapies.
Definitions
Stem cells can be divided into those that are isolated from fetal transient embryonic cell populations and those from adult tissue (adult stem cells). The former are typically isolated from blastocysts and exhibit pluripotency, meaning they can give rise to virtually any adult tissue cell type under the appropriate conditions. Adult-derived stem cells typically reside in adult tissues in a quiescent, undifferentiated state and, under appropriate stimuli, will divide and differentiate into the cell type of the tissue in which they reside or, if appropriately stimulated, into other cell types. The mechanism whereby an undifferentiated, quiescent stem cell can give rise to a multitude of differentiated, postmitotic cell types is an area of active investigation, and large-scale genomic analysis2 and transcriptional profiling3 of stem cells have led to varying definitions of “stemness.”4 While truly pluripotent embryonic stem cells (ESC) have been identified that can give rise to a multitude of differentiated cell types, there is controversy as to the generality of this phenomenon with regard to the plasticity of adult stem cells; implanted adult tissue-derived “stem cells” can, indeed, contribute to tissue regeneration in a number of different systems, but it is not entirely clear whether this is a result of a stem cell differentiating into the end cell product or if this occurs by somatic cell fusion between the “stem” cell and a population of differentiated, postmitotic, tissue-specific cells that then share the stem cell’s proliferative capabilities as well as their own tissue-specific phenotypic characteristics.
There are many examples of fetal tissue-specific somatic stem cells being identified as pluripotent cells followed by studies questioning the validity of the finding. This is particularly well illustrated by the controversy surrounding the issue of whether or not fetal neural stem cells can differentiate into hematopoietic cells; several reports describe hematopoietic competency in brain-derived neural stem cells.5,6 Shortly after this, others reported that such competency was a rare event and may depend on genetic or epigenetic alterations, not true “stemness.”7 Most recently, pluripotency, including the ability to form hematopoietic cells, was once again attributed to neural stem cells.8 Given the current lack of precision in defining how cells “choose” their phenotypic fates and activate or deactivate developmental programs, it remains unclear precisely which stem cells are actually present in somatic tissues of the fetus and adult. These issues are critically discussed in reviews to which the reader is referred.9,10,10a
Embryonic stem cells
ESC are derived from a transient population of cells in the early embryo and are characterized by pluripotency (i.e., the ability to differentiate into derivatives of the three primary germ cells layers: ectoderm, endoderm, and mesoderm) and the ability to self-renew indefinitely (Fig. 35.1). Murine ESC were first derived and cultured in 1981,11,12 and have become important research tools for the creation of transgenic and knockout mice and for the study of early development in mammals (Table 35.1).13 In 1995, James Thomson’s team derived primate ESC from rhesus monkey blastocysts,14 but it was not until 1998 that human ESC (HESC) were derived and cultured from the inner cell mass of human blastocysts15 or from human primordial germ cells.16 Human blastocysts develop approximately 5 days postfertilization and the inner cell mass consists of 20–50 cells at days 5 and 6.17 In 2006, a method for culture of HESC from single blastomeres without destruction of the embryo was reported.18 Methods for the culture of murine and HESC typically utilized fibroblast feeder cells to facilitate the growth and survival of the stem cells but recently protocols have been developed for the feeder-free and serum-free culture.13
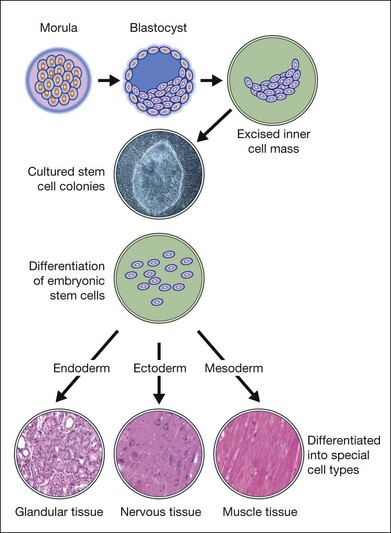
Table 35.1 Embryonic stem cell (ESC) time lines
| 1981 | Mouse ESC first derived |
| 1995 | Primate ESC isolated and grown in culture |
| 1998 | Human ESC isolated and grown in culture |
| 2007 | Differentiated adult cells reprogrammed to ESC-like cells (iPSC) |
| 2009 | First human ESC clinical trial approved by US FDA for spinal cord injury |
| 2010 | FDA approval for phase I/II clinical trial to treat Stargardt disease |
| 2011 | FDA approval for phase I/II clinical trial to treat advanced dry age-related macular degeneration |
iPSC, induced pluripotent stem cells; FDA, Food and Drug Administration.
The National Institutes for Health (NIH) have developed guidelines to establish policy and procedures under which NIH will fund research using HESC, and to help ensure that NIH-funded research in this area is ethically responsible, scientifically worthy, and conducted in accordance with applicable law. These guidelines were developed in response to Executive Order 13505, were issued on March 9, 2009, and became effective on July 7, 2009. As of November 2011, there were 136 HESC lines eligible for use in NIH-supported research, as listed in the NIH Human Embryonic Stem Cell Registry (http://stemcells.nih.gov/research/registry/).
Induced pluripotent stem cells
One of the most significant advances in the field of stem cell biology came with the observation by Yamanaka and colleagues that the addition of four critical transcription factors, under appropriate conditions, to adult somatic cells (e.g. skin keratinocytes or fibroblasts) would generate an induced pluripotent state from which a number of differentiated cells types could then be derived18a. By generating such induced pluripotent stem cells (iPSC), it may now be possible to produce autologous grafts for use in treating a variety of diseases, including those of the retina. This is discussed in greater detail below.
Adult tissue stem cells
The concept that adult tissues contain pluripotent stem cells that can serve as a source of regenerative tissue useful in the repair of aging adult organ systems is an important scientific advance.19 For example, tissues traditionally thought of as not having regenerative capacity (e.g., brain and myocardium) are the ones most often touted as being the most likely to benefit from treatments with neural or myocardial “stem cells.” In fact, clinical trials using adult bone marrow-derived stem cells to regenerate infarcted myocardium have reported success in improving cardiac function,20 presumably on the basis of bone marrow stem cells differentiating into myocardium. Unfortunately, the idea that adult bone marrow-derived hematopoietic stem cells (HSC) can transdifferentiate into myocardial tissue is not supported by experimental data.21,22 While this does not invalidate the clinical observations, it does raise a cautionary note on how such results can be interpreted and emphasizes the need to establish rigorous standards by which such clinical studies are evaluated.23
While there is an extensive literature on stem cells giving rise to nervous,24 muscle,25 vascular,26,27 and hematopoietic tissue, work on retinal stem cells is more limited. Nonetheless, a literature has emerged over the past decade that strongly supports the potential for exploiting progenitor cells to maintain, and perhaps regenerate, abnormal retinal tissue. These studies describe four basic populations of cells that may contain dormant progenitor cells which, under appropriate circumstances, may have therapeutic application in the treatment of retinal disease: (1) retinal stem cells that can give rise to photoreceptors and other retinal neurons; (2) Müller/glial stem cells that can differentiate into retinal neurons; (3) retinal pigment epithelial (RPE) stem cells that can not only serve to replace diseased RPE but perhaps also be stimulated to differentiate into photoreceptors; and (4) endothelial progenitor cells (EPC) that can contribute to the retinal vasculature and exert a neurotrophic effect. Since there are a large number of reviews on retinal stem cells, this topic will not be discussed in great detail here. Glial and RPE stem cell biology are relatively new areas of investigation and, thus, there is not yet a lot of data on this potentially exciting, but understudied, area. Adult bone marrow-derived HSC containing EPC is an area of recent interest with broad-ranging potential application and will be discussed in slightly more detail given the therapeutic potential of these cells.
Retinal stem and müller/glial cells
It has long been known from classic studies in developmental biology that the retina of amphibians and chick embryos regenerates after injury and that this regenerative capacity derives from quiescent stem cells that reside in the adult retina of these species.28,29 Given that such potential exists in lower vertebrates, there have been numerous efforts to demonstrate similar regenerative capacity in the mammalian retina. For a population of retinal stem cells to exist in the adult retina, it would be necessary for such a population of progenitor cells to remain quiescent after the retina has fully differentiated. Thus, a better understanding of gene expression during retinal development and an analysis of the orderly progression of such gene expression during the establishment of the highly ordered and regulated adult retina would provide a rationale in the search for adult retinal progenitor cells. Studies using large-scale genomic analysis30 or serial analysis of gene expression in combination with in situ hybridization31 to localize gene expression temporally and spatially to individual retinal cell types has provided such a “molecular atlas.” This work can serve as the starting point for the evaluation of numerous genes and their potential role in the regulation of retinal cell developmental determination. How genes are progressively switched on and off in an orderly fashion during the generation of specific retinal cell types, and how this overlaps with gene sets utilized during the establishment of other, nonretinal neuronal cell types, will contribute significantly to our understanding of retinal progenitor biology. The regulation of cell proliferation32 and the role of various transcription factors and signaling molecules33 during this process have provided insight into putative mechanisms whereby the mammalian retina holds in reserve a subset of progenitor cells that theoretically could be used to regenerate damaged tissue in the adult. Clearly, the developmental state of the retina and the context in which a specific cell finds itself will determine how a particular retinal progenitor cell behaves; whether it will terminally differentiate or maintain a quiescent state from which it can later emerge to give rise to cells useful in the repair of a damaged retina will depend not only on its own developmental program, but the microenvironment in which it finds itself.34 Transcriptional profiling studies of retinas at different states of development coupled with in vitro studies of progenitor cell populations35 should provide the information necessary to begin such an analysis and determine what conditions help maintain quiescence and what conditions stimulate proliferation and subsequent differentiation of retinal progenitor cell populations.
Cells with characteristics of retinal neurons have been obtained from a number of embryonic tumor cell lines, including neuroblastoma, glioblastoma, and teratocarcinoma,36 but given the malignant potential of these cells, are not likely to be useful in the treatment of retinal degenerative disorders. In subsequent studies, cultured virally transformed, immortalized neuronal precursors were evaluated for their ability to differentiate into retinal neurons after intraocular implantation.37 Attempts at inducing differentiation of ESC into retinal neuronal phenotypes were made using a variety of factors, including retinoic acid.38 It has been reported that retinal progenitor cells can, indeed, be identified in the adult retina. Such cells exist in the ciliary margin; single pigmented epithelial cells can be isolated from the ciliary margin (but not the central or peripheral pigmented epithelium) and clonally expanded in culture to give rise to a variety of retinal cell types, including rod photoreceptors, bipolar neurons, and Müller glia.39,40 The differentiation of these cells from the ciliary margin pigmented cell progenitors is not due to transdifferentiation of the ciliary margin cells, but, rather, clonal proliferation and differentiation, as observed in a true stem cell. Thus, the potential utility of these cells and their characterization have come under scrutiny and are discussed extensively in a number of excellent recent reviews.41–44
In lower vertebrates (e.g., chickens) adult, differentiated Müller glia themselves can serve as a source of stem cells that will, in response to injury or exogenously added cytokines, dedifferentiate, proliferate, and redifferentiate into additional glial cells or neurons.45 A common progenitor cell that gives rise to both Müller glia and retinal neurons was described nearly two decades ago.46 Thus, the concept that Müller glia of the adult retina can serve as a potential source of retinal stem cells is consistent with molecular profiling of developing mammalian retinas that shows a high degree of similarity between the gene expression profiles of Müller glia and mitotic retinal progenitor cells in the mouse.31 Since the Müller glia are the cells that commonly proliferate in response to retinal injury, it would not be surprising that these cells also retain the potential to differentiate along a number of pathways, some of which may lead to retinal neuronal replacement. One study has significantly expanded this concept significantly: amacrine, horizontal, and photoreceptor phenotypes were expressed by Müller glial cells following toxic injury to the adult mammalian retina in the presence of extrinsic factors (e.g., retinoic acid) or activation of intrinsic genes.47 These studies are most provocative, could provide additional insight into retinal regeneration in mammals, and provide a rationale for the targeting of Müller glia in certain inherited and acquired retinal degenerative disorders.
Müller glia are the only cells that span the entire neurosensory retina; cell processes extend anteriorly to the ganglion cell layer as well as posteriorly to the RPE and ramifications of these processes form intimate contacts with retinal blood vessels, photoreceptors, and other retinal neurons. Müller glia are activated in response to vascular changes such as those observed in a variety of retinal vascular and neurodegenerative diseases.48,49 Upon activation, these cells upregulate glial fibrillary acidic protein and in animal models of outer retinal neovascular disease their appearance and location precisely correlate, both temporally and spatially, with subretinal neovascularization and associated neuronal degeneration in the outer retina.50 Recent studies describe the use of activated Müller cell targeted adeno-associated viral vectors containing a transgene encoding a neurotrophic molecule to target the outer retina in vasculodegenerative50 and neurodegenerative disorders51 characterized by photoreceptor degeneration. This strategy using endogenous cells (e.g., activated Müller cells) to deliver gene therapy products to the outer retina could be useful clinically to avoid the need for subretinal injections of the viral vector, a procedure that can have deleterious effects on already diseased retinas.
Differentiation of HESC into photoreceptors
Studies evaluating the in vivo efficacy of HESC-derived photoreceptors are at an early stage of development. Differentiation of HESC into retinal progenitor cells has been achieved, as has further in vitro differentiation into photoreceptor-like cells.52 As compared to RPE, photoreceptor transplants would require integration into the existing outer nuclear layer and formation of functional synapses to act as a replacement therapy. Conversely, there may be some potential therapeutic effect by providing neurotrophic support to adjacent cells without true synaptic integration. When HESC-derived retinal progenitor cells were cocultured with explants from retinal degeneration mice (Aipl1–/–), they showed incorporation into the retinas, had morphologic characteristics of photoreceptor cells, and were immunoreactive for recoverin – a finding that was only rarely found when cells were cocultured with wild-type retinas.52 Subsequent experiments showed that when these cells were injected into the subretinal space of adult Crx–/– mice (a model of Leber congenital amaurosis), the HESC-derived retinal cells differentiate into photoreceptor-like cells that were immunoreactive for recoverin and rhodopsin and restored light responses with an electroretinogram-like signal in the transplanted eye.53 While the integration of HESC-derived photoreceptors may be a relatively rare event, these results demonstrate in principle that HESC can be used as a source of cells for photoreceptor replacement therapy.
Studies have shown that Noggin (inhibitor of BMP pathway) or Dickkopf (Dkk)-1 (an antagonist of wnt signaling pathway) promote anterior neural identity and then insulin-like growth factor (IGF)-1 promotes the formation of retinal progenitor cells.54 Tom Reh’s lab reported derivation of retinal and photoreceptor-like cells from HESC-derived embryoid bodies grown in suspension culture, through addition of Noggin, Dkk-1, and IGF-1 into neuronal differentiation medium in adherent culture.52,55 After 21 days of induction in the differentiation medium, they found the expression of retinal progenitor cell-related transcription factors such as Rx, Otx2, Pax6, Chx10, and Crx was significantly increased.55 When further cultured, these cells formed neural rosettes that were picked and, allowed to self-aggregate, they formed rosettes with retinal progenitors in the center, differentiated cells showing photoreceptor markers such as recoverin, and RPE differentiation at the periphery.55 Microarray analysis of HESC-derived retinal cells showed a very high correlation between genes expressed in human fetal retina and HESC-derived retinal cells.56
Investigators from the RIKEN Research Institute reported the successful derivation of retinal progenitor cell and photoreceptors from either mouse or HESC using an even more highly defined stepwise method also using suspension culture followed by adherence culture.57,58 The final steps included use of retinoic acid and taurine to induce photoreceptor differentiation. With this induction method, they found that HESC can be differentiated into photoreceptors showing both rhodopsin and recoverin immunoreactivity in 150 days.58
Differentiation of HESC into three-dimensional embryonic and retinal tissues
Murine ESC can be differentiated into eye-like structures with cells that have properties of the crystalline lens, neural retina, and RPE.59 Dissociated cells from these eye-like structures were shown to integrate into the retina, especially after retinal injury.60 In April 2011, investigators reported the remarkable discovery that murine ESC aggregates grown in presence of added basement membrane components formed hollow spheres that underwent evagination into vesicles and then invagination into optic cup structures. The self-organizing optic cups had RPE and neural retinal domains and generated stratified neural retinal tissues61 (Fig. 35.2). Later in 2011, it was reported that three-dimensional populations of human retinal progenitor cells can be isolated from early forebrain cultures derived from HESC, and that a transient population of optic vesicle structures arose during the time appropriate for human retinogenesis. These optic vesicle structures could be differentiated into multiple retinal cell types, including RPE and photoreceptors.62
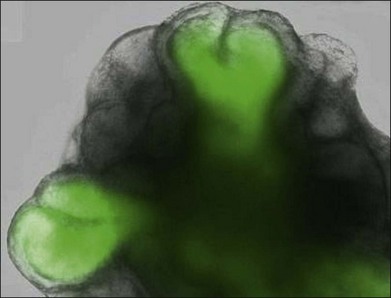
Clearly, there would be great therapeutic value in isolating retinal stem cells from the adult mammalian eye and then using them to regenerate diseased retinas. Recent reports suggest that, if done early enough, some level of visual function may be obtained in animal eyes with retinal degeneration treated with multipotent progenitor cells isolated from retinas of neonatal mice.62a
Retinal pigmented epithelial stem cells
Transplantable RPE cell lines may serve as stem cells of sorts to replenish diseased RPE cells themselves. In a number of macular and retinal degenerative disorders there is atrophy of the RPE and associated malfunctioning in the phototransducing cellular machinery. Damaged RPE cells and associated atrophy are hallmarks of age-related macular degeneration (AMD) and heroic surgical approaches63 have been considered to provide photoreceptors in such individuals with healthier, RPE-rich regions of the retina through retinal translocation and the insertion of RPE sheets. While the former has enjoyed a limited clinical effort,64 the latter is still in the laboratory stages of development and has not yet been employed in the clinic.65,66
RPE cell-based delivery of trophic (and other) factors
Immortalized human RPE cell lines have been created to facilitate RPE cell transplantation as well as their use in cell-based therapeutic approaches. These cells enjoy an extended lifespan after being stably transfected with a plasmid encoding the simian virus 40 large T antigen and many of the factors expressed by functional RPE cells in vivo are observed to be expressed by these transformed cell lines.67 When these cells are transplanted subretinally into a rat model of retinal degeneration (the Royal College of Surgeons (RCS) rat), loss of visual function is attenuated68 and cortically dependent visual function is preserved long-term.69 These RPE cell lines can be transfected with plasmids encoding a variety of trophic factors shown to have protective effects on photoreceptors70,71 and then encapsulated into polymer devices that permit diffusion of cell products into the tissue into which they are transplanted. When transformed RPE cell lines are transfected with a plasmid encoding one such factor, ciliary neurotrophic factor (CNTF), and transplanted directly into the vitreous of dogs with retinal degeneration, photoreceptor degeneration is reduced.72 Furthermore, production of this factor and implantation of the encapsulation device into the vitreous of normal rabbits did not lead to toxic effects on either the electroretinogram or retinal histology in these animals at doses that protect photoreceptors in dogs with retinal degeneration.73 Such cell-based delivery devices may be used to provide trophic factors for the treatment of a variety of retinal degenerative diseases and, in fact, have recently been used in a human clinical trial evaluating the efficacy of CNTF in the treatment of retinal degeneration.73a In this respect, the implanted encapsulated cell devices function as a stem cell might, providing factors critical to the prevention of, or recovery from, retinal degenerative disease.
Embryonic stem cells as a source of RPE
Over the past decade, a number of methods have been reported to achieve the differentiation of HESC into retinal cells.52,58,74–76 Some of these methods were developed for differentiation of HESC into one type of retinal cell such as the RPE58,74–76 or photoreceptors,58,75,76 while other methods have focused on the efficient generation of retinal progenitor cells.52
Differentiation of HESC into RPE
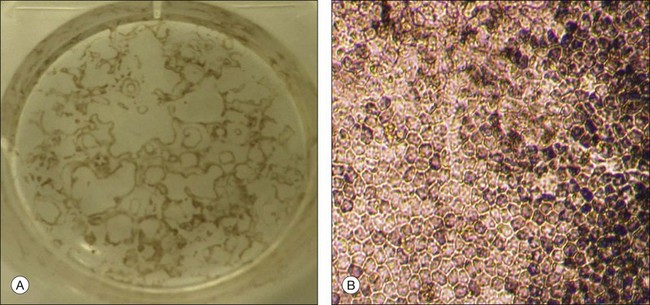
Spontaneous differentiation of HESC into RPE is the simplest and most commonly used method to produce RPE from HESC.75 HESC colonies are first allowed to overgrow in growth factor-supplemented HESC medium until the borders of the colonies contact each other. The medium is then changed to basic HESC medium without basic fibroblast growth factor supplementation and is changed every other day for several months until the RPE cells are seen as small pigmented colonies in the culture dish (Fig. 35.2). RPE can also be derived from HESC using a two-stage induction method: HESC are first differentiated towards a neuroectodermal fate in suspension culture using neural differentiation medium, and then differentiated into RPE. The neural precursors are then plated in cell culture dishes for further differentiation into RPE. During these stages of induction and differentiation, the RPE cells appear as early as 4 weeks and reach a large enough number of cells for subculture at approximately 8–10 weeks.75 Nicotinamide and Activin A (a member of the transforming growth factor-beta superfamily) can also be used to direct the induction of RPE from HESC.83
RPE cells can be easily identified and isolated from other differentiated cells in HESC cultures because of their unique pigmentation, hexagonal shape, and pattern of growth. The RPE patches can be either mechanically incised out of the cultures or enzymatically dissociated from the cultures and can be grown to confluence and passaged retaining typical pigmentation and morphology (Fig. 35.3).
Characterization of HESC-derived RPE in vitro
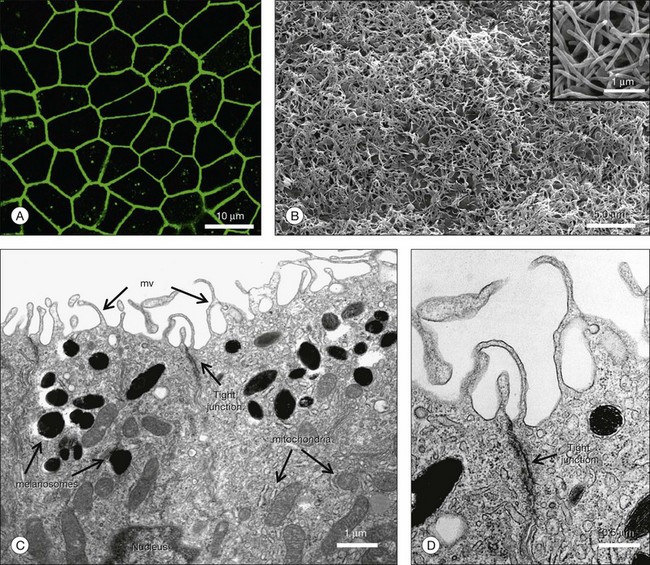
The characteristics that define human RPE cells have been outlined in Chapter 16 (Cell biology of the retinal pigment epithelium) and reviewed by others.74 HESC-derived RPE develop a typical hexagonal shape and become highly pigmented when they attain confluence (Fig. 35.3B). The cells can further differentiate and become highly polarized when grown for extended periods of time on Transwell inserts and other substrates (Fig. 35.4).77,78 Polarized HESC-derived RPE show apical microvilli, are joined in the apical regions by tight junctions, show apically distributed sodium/potassium ATPase (Na+/K+ATPase), and have a high transepithelial resistance.78 HESC-derived RPE express a host of RPE-specific and RPE highly expressed genes including visual cycle genes (RPE65, RDH 11, CRALBP); RPE membrane channel and transporter genes (BEST1, SLC); pigment biosynthesis and melanin biosynthesis genes (GPR143, TYRP1, dopachrome tautomerase (DCT), SILV); and phagocytosis-associated genes (LAMP2, VDP, Mertk, GULP1).75
Efficacy of HESC-derived RPE cells in vivo
The most commonly used model to evaluate therapeutic efficacy of HESC RPE is the RCS rat. The primary defect in the RCS rat is in the RPE; thus this model provides the ability to evaluate the effectiveness of RPE cell replacement therapy. The RCS rat has a recessively inherited mutation in the receptor tyrosine kinase gene Mertk, leading to impaired phagocytosis of shed photoreceptor outer segments with buildup of outer-segment material in the subretinal space, and subsequent secondary degeneration of photoreceptors between postnatal days 20 and 60.79 Using subretinal injections of cell suspensions of HESC RPE, several groups have shown reproducible survival of the transplanted HESC RPE in the subretinal space (>220 days in one study), and positive labeling of these cells with human-specific markers and RPE-specific genes (e.g., RPE65).80–83 The cells appear to disperse in the subretinal space and, while clumps of cells or multilayered grafts are often formed, in some cases they were arranged in an apparent monolayer. Cells showed focal rhodopsin staining, suggesting that they are phagocytosing photoreceptor outer segments.81–83 The transplanted HESC RPE were associated with histologic and functional rescue of the photoreceptors, as measured by delay in the loss of nuclei in the outer nuclear layer, and retention of electroretinogram and optomotor responses in the treated eye compared to untreated eye.80–83 Each of these studies utilized immune suppression (typically systemic cyclosporine, with or without systemic corticosteroid) to prevent immune rejection of the xenograft. Although the subretinal space is thought to be an immune-privileged site, such privilege may be compromised at the time of surgery or by the disease process.84,85
Pivotal safety studies are necessary prior to receiving approval from the Food and Drug Administration (FDA) for a clinical trial. Studies using HESC-derived RPE cell suspensions under good manufacturing practices and good laboratory practice have been performed to establish karyotypic stability in the cells, lack of infectious and adventitious agents in the product, and lack of teratoma and/or tumor formation by the cells in immune-deficient mice.81 It should be noted that G-banding karyotypic analysis may not show any significant abnormalities while high-resolution DNA analysis may show culture-induced copy number changes and loss of heterozygosity; the significance of these changes for the purpose of cell therapy is unknown.18 Importantly, none of the studies that utilized highly differentiated HESC-derived RPE in immune-deficient animals found evidence of teratoma formation.80–83 However, one study in which murine ESC differentiated toward RPE fate for only 7 days and injected in the subretinal space of rd12 (a spontaneous mutant model of Rpe65 Leber congenital amaurosis and having the same genetic background as the murine ESC) developed teratoma tumors in some animals.86
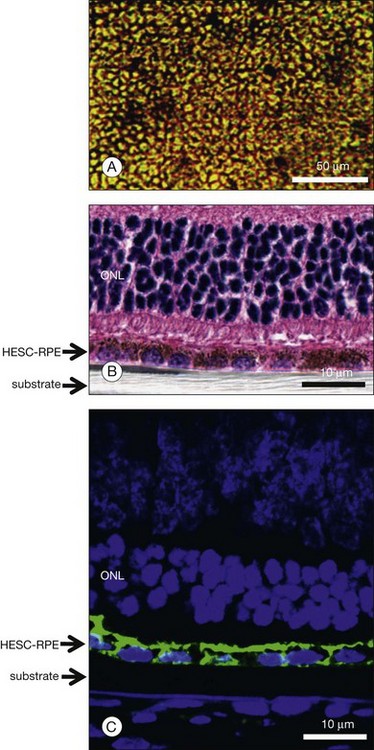
An alternative approach to the use of HESC RPE cell suspensions would be to transplant monolayer sheets of highly differentiated and polarized RPE resting on a biodegradable or nonbiodegradable membrane. The reasoning for such an approach would be that these cells would have a better chance to integrate with the host photoreceptor outer segments as an intact monolayer and thus improve the functionality of the graft.77 It has been shown that HESC RPE can be polarized in vitro to develop tight junctions with high transepithelial resistance and elaborate extensive apical microvilli (Fig. 35.4).78 It has also been observed that highly polarized HESC RPE show increased secretion of pigment epithelial-derived growth factor, a factor with neurotrophic and antiangiogenic activity.78 HESC RPE are capable of specifically phagocytosing photoreceptor outer segments.86a Polarized HESC RPE show increased phagocytosis of bovine rod outer segments in vitro compared to nonpolarized cultures.78 Studies implanting polarized HESC RPE grown on a nonbiodegradable substrate (e.g., parylene) show retention of an intact monolayer in vivo and prominent integration with host photoreceptors (Fig. 35.5).77 The relative efficacy of HESC-derived RPE cell suspensions versus polarized sheets is under active investigation by several groups.
The effect of the local tissue microenvironment on transplant survival is an important issue when considering how best to deliver stem cells and cells derived from stem cells. Cross-talk between cells and extracellular matrix is critical to maintaining the differentiated cell type as well as insuring appropriate function. Many studies, in the retina as well as other tissues, have identified a number of factors that may be critical to the successful engraftment of stem cell-derived, and other, cell types, including cardiomyocytes,87 spinal cord,88 and brain89 neurons and photoreceptors.90
The use of induced pluripotent stem cells as a source of autologous RPE (and other cell type) grafts
One of the most significant recent breakthroughs in stem cell research has been the observation that adult human somatic tissues may be induced to express pluripotency after reactivation of four transcription factors.91 These adult tissue-derived stem cells, called induced pluripotent stem cells (iPSC), can be differentiated into a variety of tissues that may be used as autologous grafts for therapeutic reimplantation in various diseases.92 The use of iPSC circumvents most of the ethical and practical problems associated with large-scale clinical use of ESC, and patients with iPSC-derived autologous transplants may not require lifelong immunosuppressive treatment to prevent graft rejection. However, safety concerns regarding iPSC include malignant transformation resulting from reactivation of the four reprogramming transcription factors (including c-MYC) randomly integrated into the genome at multiple loci following retroviral transduction. In fact, iPSC generation and transformation share many molecular mechanisms, and a high incidence of tumorigenesis is observed in iPSC-derived chimeric mice.93–96 The risk of tumorigenesis cannot be attributed solely to c-MYC reactivation: transgenic mice derived from iPS without c-MYC still form tumors.97
Exhaustive comparative analyses of multiple human (h) iPSC and HESC lines reveal that, while many hiPSC and HESC lines share very similar transcriptomic and epigenetic profiles, others are heterogeneous. The differences observed are randomly distributed and limit the differentiation capacity of the cells.98 Furthermore, recent evidence shows that, in hiPSC, reprogramming and selection pressure to obtain rapidly proliferating cell-lines may induce chromosomal aneuploidy in nonrandomly distributed loci that may further limit the differentiation capacity and promote tumorigenicity of iPSCs.99–101 Genetic instability in iPSC is correlated with higher passage numbers, so reprogramming methods that are inefficient and require multiple passages may increase the risk of tumorigenesis.99,100 Furthermore, high cytotoxicity is observed when reprogramming methods that utilize repeated transfections (reprogramming methods involving mRNA strands or recombinant proteins). Consequently, these methods could induce even higher selection pressures for highly proliferative cells with mutations; however this idea has not yet been directly tested.
To generate RPE from hiPSC for treating diseases such as AMD in which the onset occurs in 55+-year-old humans, the source material (likely fibroblasts or keratinocytes) will be older and more difficult to reprogram. The RPE derivation process is long and all clones will need to be carefully screened before transplantation. Consequently, this treatment strategy could be very economically taxing to patients and healthcare systems. Therefore, to generate any tissue of interest, including RPE from hiPSC, one or two specific reprogramming protocols may need to be adopted and optimized to insure reliable and safe derivations of that cell type. Furthermore, experimental animals with implants should be monitored over extended periods of time to ensure that no tumors form. We have recently reported the generation of iPSCs reprogrammed using two or one factors (OCT4/KLF4 or OCT4 only) and small molecules.102,103 Recent advances in stem cell biology have demonstrated that iPSC can be derived from somatic cells such as skin fibroblasts by using retroviral vectors to introduce reprogramming factors (Oct4, Sox2, Nanog, Lin28) into somatic cells. However, random viral integration and spontaneous reactivation of reprogramming factors during differentiation pose a cancer risk and limit the potential use of iPSC derived in this manner in human therapy. In addition to this risk, therapeutic application of current technology is also limited by its low efficiency and slow kinetics. Finally, there is the requirement for numerous growth factors and animal cell feeder layers to obtain even limited numbers of iPSC. To overcome these hurdles, increase the efficiency of generating iPSC-derived RPE, and avoid using lentiviral vectors, it may be possible to use an episomal, or nonintegrating, vector to deliver the required transcription factors or eliminate the need to deliver such factors by using small molecules to induce pluripotency.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


