2 The approach to successful diagnosis and therapy begins with the clinical history. The medical interview should serve to collect information helpful to detect, treat, and prevent illness. Obtaining the history requires considerable skill that is developed only through extensive experience. As physicians, we should remember to perform thorough but goal-oriented questioning of each patient. Many patients fail to mention details relevant to the history or symptoms that affect them because they forget or think there is no relation to their eye problem. During the dialogue, patients will provide a more accurate testimony if they are comfortable and good rapport has been established between them and the clinician. A careful interrogation of the patient regarding his or her past history and chief complaint can produce a broad clinical picture that leads to the suspicion of different categories of retinal disease (Table 2–1). Information obtained from the history frequently leads us to pay particular attention to subtle aspects of the ocular or physical examination. The patient’s history is also used to guide the diagnostic workup that eventually may point in the direction of a particular retinal disease. A fact that is applicable to every patient who has a visual or ocular complaint is that a carefully obtained history makes reaching a correct diagnosis more likely. Although often diagnoses and treatment decisions of retinal pathology rely in large part on a meticulous ocular examination, in a significant percentage of cases, sorting out the proper retinal diagnosis may depend largely on the history.
Sorting Out Possible Diagnoses from the History
What Is the Importance of the Clinical History?
| Age |
| Gender |
| Race |
| Place of birth and history of travel and residence |
| Place and type of employment |
| Family medical and ocular history |
| Past medical and ocular history |
| Medications |
| Diet and habits |
| Review of systems |
| Chief complaint and history of present illness |
What Elements in the History Are Useful in Sorting Out the Diagnosis?
Age
This is probably the most important piece of demographic information leading to the inclusion or exclusion of large numbers of probable diagnoses. The retinal diseases that commonly affect the newborn are rare for older children. Retinal disorders frequently encountered in older patients differ from those found in middle-aged patients. There are many reasons for the effect of age on retinal pathology. Genetic defects that affect ocular physiology manifest more commonly in the earlier years of life; susceptibility to infectious insult is higher at the extremes of our life term; and time-related degenerative changes lead to disease in older persons. A clear example of age affecting the selective suspicion for retinal diagnoses is age-related macular degeneration (AMD) of older patients, which contrasts with inherited macular dystrophies, which are much more typical in young and middle-aged persons. Retinoblastoma, leukemia, Coats disease, and familial exudative vitreoretinopathy are most often retinal diagnoses of childhood. Birdshot and serpiginous chorioretinopathy, in contrast, are more prevalent in persons older than 50 years of age. Masquerade syndromes should be suspected at both ends of the spectrum. Metastatic carcinoma, uveal melanoma, intraocular lymphoma, ocular ischemia, and paraneoplastic retinal syndromes usually occur in older persons.
Gender
Many eye diseases are more common in one gender than the other. Multiple facts explain this, including retinal diseases that may be linked to hormonal factors or those that are linked to a gene defect in a sexual chromosome and other less understood reasons. Central serous chorioretinopathy, for example, affects men much more frequently than women. Incontinentia pigmenti, on the other hand, is found almost exclusively in women.
Race
The ethnic background of a patient is very pertinent information when discerning possible retinal diagnoses because many disorders have marked differences in their incidence among various races. These differences are probably due to certain gene defects and other forms of molecular predisposition to disease being found more commonly in one race than in others. For instance, birdshot retinochoroidopathy in white persons has a strong association with antigen human leukocyte antigen (HLA)-A29. Another example is the case of melanin in uveal tissue, a proposed protector against environmental light irradiation. Its relative abundance or scarcity is believed to predispose for different ocular pathology: Vogt–Koyanagi–Harada disease is an autoimmune affliction against melanin in darker-skinned patients, usually Asians, Hispanics, and Native Americans; uveal melanoma has a correlation with sun exposure in less pigmented persons, such as those of Northern European ancestry. Diseases like sickle cell disease retinopathy or sarcoid uveitis occur much more frequently in African Americans. Other disorders, like white-dot syndromes, inflammatory chorioretinopathies, or AMD, have a much higher prevalence in the white population.
Place of Birth and History of Travel and Residence
A patient’s geographic history holds important in the consideration of retinal conditions, particularly those of an inflammatory nature. Ocular infectious agents and environmental factors that affect them often are limited to certain locations and climates. This is the case for presumed ocular histoplasmosis syndrome or fungal and parasitic diseases, including onchocerciasis. Socioeconomic factors, dietary customs, and hygienic practices may vary in different places, again increasing the risk for infectious retinal diseases such as toxoplasmosis, which is associated with poor sanitation and the practice of eating undercooked meat.
Place and Type of Employment
External risk factors for retinal disease may be linked to the type and location of the occupation of a patient. Exposures to potential toxic agents, trauma resulting in a rhegmatogenous retinal detachment, and excessive sun exposure related to uveal melanoma are possible associations between employment and retinal disorders. Patients who work in sewers may be in contact with rodent urine and therefore at risk for leptospirosis, whereas brucellosis is more likely to occur in farmers and veterinarians.
Family Medical and Ocular History
This part of the history is particularly important in considering inheritable retinal diagnoses. Some retinal diseases are autosomal dominant and have a classic mendelian inheritance pattern. A positive family history may orient to conditions such as vitelliform dystrophy or retinitis pigmentosa, for example. Other inheritable retinal disorders are recessively transmitted: The presence of disease in siblings or history of consanguinity may suggest the diagnosis, as it may occur in gyrate atrophy or fundus flavimaculatus.
Past Medical and Ocular History
Of all the mentioned components in the history, prior history of disease, either systemic or ocular, has the highest yield in suggesting the cause of the complaints the patient describes. Complete past medical and surgical history often will unveil systemic illnesses associated with retinal diseases. Many common systemic diseases have a retinal component. The most familiar scenarios are diabetes mellitus, systemic hypertension, and rheumatic diseases. History of previous ocular disease is naturally very relevant to possible retinal complications or new disorders that a person may develop. An example is high myopia in a patient complaining of progressive central loss of vision, in which the suspicion for choroidal neovascularization is very high. Another common situation is endophthalmitis in a patient who recently had intraocular surgery.
Medications
Commonly used systemic medications have been associated with retinal disease. Medications well known for resulting retinal degeneration include chloroquine, hydroxychloroquine, and thioridazine. The interferons need to be monitored for the retinal vascular occlusive disease they often induce. Corticosteroids are an important consideration when central serous chorioretinopathy is suspected.
Diet and Habits
Avitaminosis, particularly among children and older persons, may cause retinal disease. Vitamin A deficiency is an example of producing fundus xerophthalmia and nyctalopia. As mentioned earlier, consumption of raw or partly cooked meat can lead to toxoplasmosis or cysticercosis. Individuals who include unpasteurized milk in their diet are at risk of tuberculosis and brucellosis. Important habits to keep in mind are drug use, such as intravenous injection of heroin, which can result in retinal arterial occlusions if the drug is impure or in endogenous endophthalmitis. Sexual promiscuity and sometimes drug use are important risks for human immunodeficiency, herpes viruses, and syphilitic uveitis. A history of contact with unwormed dogs is significant in children with suspected ocular toxocariasis.
Review of Systems
For many retinal diseases, a meticulous review of systems is necessary to establish the diagnosis. It serves to recognize relevant symptoms previously unrecognized by the patient to be relevant to his or her eye illness. This is particularly true in uveitic, vascular, or neoplastic retinal disorders. An example is HLA-B27-associated uveitis, which frequently coincides with pathology in the skin, mucosae, joints, and gastrointestinal system. Unless directly asked, patients do not realize that there may be a common source with their uveitic disease, which is why they usually fail to mention complaints affecting other organs. Recognition of systemic symptoms is not only extremely helpful in arriving at the retinal diagnosis, but it can be used to prevent serious progression of disease elsewhere in the body.
Chief Complaint and History of Present Illness
These categories of information in the clinical history contain the key facts needed to reach a probable diagnosis. For this reason, the physician seeks these components in the history first in the interrogation. The chief complaint is the main reason the patient has come to the ophthalmologist. Identification of the chief complaint allows the physician to recognize the most relevant symptom of the patient’s disease and immediately raises certain categories of retinal disease versus others. In the history of present illness, the severity, time of onset, course, and other characteristics of the patient’s retinal symptoms, starting with the chief complaint, must be thoroughly described. Blurred or decreased vision is most commonly the symptom for which patients seek care. Other usual symptoms include scotomata, which suggests focal retinal damage; metamorphopsia, which can result from mechanical distortion of macular topography from conditions such as epiretinal membranes or sub-retinal or intraretinal fluid; and flashes and floaters, which are common in vitreoretinal traction syndromes. Pain and photophobia, when associated with retinal disease, are symptoms that most commonly reflect anterior intraocular inflammation affecting the iris or ciliary body or elevated intraocular pressure.
How Should the Information Be Included in the Patient’s History of Present Illness?
The history of current illness is a chronologic description of the patient’s complaints. Each symptom should be distinguished by its type and quality; its intensity; time of onset, and mode of progression; its duration and frequency of recurrence; the existence of precipitating factors, alleviating factors, or aggravating factors; and associated events that precede, occur simultaneously, or follow the symptom described. Retinal diseases typically present with visual and ocular symptoms, which often can be explained on the basis of the underlying pathophysiologic process. One type of information that is particularly revealing in retinal problems is whether the complaint is monocular or binocular and to what extent symptoms may be asymmetric. Important clues to retinal diagnosis, particularly those of an inflammatory nature, also may lie in systemic complaints, including those that affect the neurologic system, the skin and mucosae, the musculoskeletal system, the respiratory and gastrointestinal systems, and others.
The clinical experience and knowledge base of the individual physician affect the process by which that physician classifies and reasons about information obtained in the history. One of the complexities affecting the practice of caring for retinal diseases, much like the rest of medicine, is that patients do not consult the physician while having a known diagnosis but seek alleviation of one or more symptoms. Symptoms in each individual patient are subjective and are modified by the particulars of the disease process itself. Not infrequently, symptoms are forgotten, suppressed, or exaggerated by patients. The ability of patients to report about the retinal disease affecting them is influenced by their age, education level, gender, cultural background, socioeconomic status, and ethnicity. Most patients with a retinal illness present with both specific and nonspecific symptoms. The former are key complaints the patient reports that point to a specific disease. Nonspecific symptoms are those that will not be decisive in reaching the diagnosis because they do not form part of a cluster of symptoms that we recognize as corresponding to a specific pathological entity. Determining the level of accuracy of reported symptoms in retinal disease is an important task. The precision of the descriptions given by the patient affects our efficiency to sort out a pattern that will lead to suspicion of a particular diagnosis.
What Does the Process of Sorting Out Possible Diagnoses Entail?
Development of a reasonable suspicion for one or more retinal diagnoses requires an analytic process that resembles the scientific method, in which observation leads to a hypothesis that can be tested with experimentation to refute or confirm the hypothesis. Physicians formulate potential diagnoses based on the clinical observations that will guide diagnostic testing, or experimentation, to hold one possible diagnosis over others.
The collection of clinical information, starting with the history, is essential to problem solving in retinal diagnosis. The application of scientific reasoning becomes a repetitive cycle during the process of sorting out one true clinical conclusion. As data are accumulated regarding a retinal illness, new hypotheses are produced and supported, and others are invalidated. The physician must remain alert for clinical clues that do not fit the current hypothesis and that might make it necessary to consider renewal of the assessment of information.
The diagnostic process in the retina, and in other fields of ophthalmology as well, is a dynamic one that begins with the history. This process is carried out not only as linear thinking, but several lines of evidence are considered in a parallel manner to reach one or more suspect diagnoses that will be investigated during the physical examination, laboratory testing, and other special testing.
What Retinal Diagnoses Are Suggested by Reduction in Central Visual Acuity?
Loss of foveal vision, even in small degree, is probably the most commonly reported symptom by patients who consult the ophthalmologist. The impact on the patient varies, depending on the type and degree of vision loss, whether onset is sudden or gradual, and also whether one or both eyes are affected. Many non-retinal causes exist for the loss of central visual acuity, including refractive conditions, diseases of the cornea and lens, glaucoma, and neuroophthalmologic disorders.
Because of the lack of anterior ocular findings, neuroophthalmic diseases often need to be carefully distinguished from retinal conditions. An important question is whether the degree of visual loss is explainable by the retinal findings. For example, a patient with mild epiretinal membrane maculopathy and severe reduction in visual acuity should be further investigated for other possible causes of visual loss. Diseases of the optic nerve, such as optic neuritis, ischemic optic neuropathy, and impinging tumors, are usually unilateral. A characteristically bilateral optic nerve pathological process that may lead to pronounced visual loss is papilledema seen in increased intracranial pressure. Chiasmal lesions can result in a visual loss that exhibits a variety of patterns with bilateral field involvement. Retrochiasmal neurologic disorders affect typically bilateral vision but spare half the macular vision without a decrease in visual acuity.
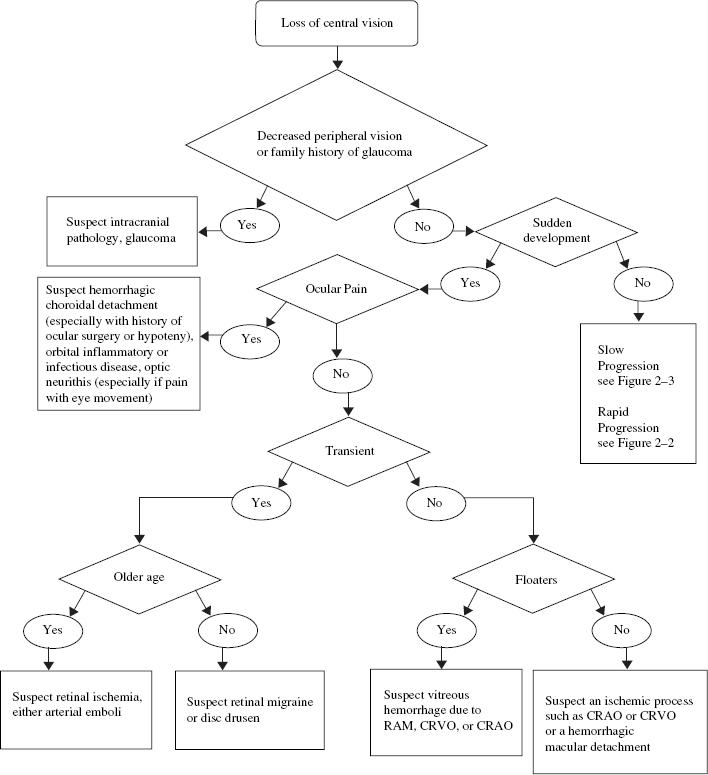
Numerous retinal causes exist for diminished foveal vision, and these causes may or may not be accompanied by a loss of peripheral vision, including ischemic processes, ocular inflammatory processes, retinal detachment, vitreous hemorrhage, retinal toxic insults, infectious disorders, dystrophic and degenerative diseases, and neoplastic disorders.1,2 A method for determining the cause of central vision loss is outlined in Figures 2–1, 2–2, and 2–3. Here we discuss central vision loss subdivided by its association with other characteristics and symptoms.
Which Retinal Diseases Are Suspected in Severe Loss of Central Vision?
Sudden and severe loss of vision is a traumatic event that usually prompts the patient to seek immediate medical attention. In older patients, especially if predisposing conditions in the medical history of the patient exist, ischemic processes leading to loss of central vision should be among the first considered. Such retinal diseases include branch retinal artery obstruction, central retinal artery occlusion, ocular ischemic syndrome, branch retinal vein occlusion, central retinal vein occlusion, and hemiretinal vein occlusion. Retinal detachment is a frequent cause of painless monocular loss of visual acuity. Severe loss of vision can occur with the three pathogenic forms of retinal detachment. Rhegmatogenous retinal detachment secondary to a retinal break often is associated with a history of aphakia, pseudophakia, ocular trauma, and high myopia. Tractional retinal detachment can be present in patients with a history of proliferative diabetic retinopathy, retinopathy of prematurity, penetrating trauma, proliferative vitreoretinopathy, angiomatosis retinae, Behçet syndrome, Coats disease, or pars planitis. Exudative retinal detachments are associated with neoplasms, Harada disease,3 posterior scleritis, collagen–vascular disease, malignant hypertension, sympathetic ophthalmia, uveal effusion syndrome, postocular surgery,4 and toxemia of pregnancy. Choroidal neovascular (CNV) membranes bleed and leak, producing painless loss of vision. In most cases, it begins as a monocular complaint. Associated conditions include neovascular AMD, presumed ocular histoplasmosis syndrome (POHS), high myopia, angioid streaks, sarcoidosis, multifocal choroiditis, punctate inner choroidopathy, melanoma, hemangioma, osteoma, and laser photocoagulation. Development of CNV membranes in patients with multiple evanescent white-dot syndrome may occur on rare occasions and also can be idiopathic in some patients.