8 The natural pigmentation of the fundus is provided by the retinal pigment epithelium (RPE), which is located between the retinal photoreceptors and the Bruch membrane. The RPE is a monolayer of hexagonal pigmented cells with the apical portion enveloping the photoreceptors and the basal aspect forming the innermost portion of the Bruch membrane. These cells contain melanosomes of neuroectodermal origin that develop during the second trimester. The RPE cells are most dense in the posterior pole, where they assume a low cuboidal configuration. The functions of the RPE layer include phagocytosis of the outer segments of the rods and cones, formation of the outer blood-ocular barrier, vitamin A metabolism, and absorption of radiant energy that is subsequently dispersed via the choriocapillaris. Changes in the RPE can be caused by a variety of mechanisms, including trauma, inflammation, retinal detachment, or congenital alterations. Hypertrophy shows an increase in the size of the RPE cells with large spherical melanin granules. This change may be congenital or acquired through aging or trauma. Hyperplasia of the RPE also displays an element of hypertrophy, but there is migration of the RPE cells and pigment into the neurosensory retina, which can give the appearance of fine bone spicules, often in a perivascular distribution. Atrophy of the RPE is the result of loss of pigmentation within these cells, which can result in a complete loss of RPE and degeneration of the overlying photoreceptors. Many of the pigmentary changes are congenital in nature. Lightly pigmented persons may exhibit a “blonde”-appearing fundus where visualization of the underlying choroidal circulation through lightly pigmented RPE is possible. A blonde fundus may also occur with autosomal-recessive oculocutaneous albinism, where there is a reduction in the amount of primary melanin deposited in each melanosome of the RPE cells. A similar fundus appearance may be observed with ocular albinism, where there is a reduction in the number of melanosomes. Ocular albinism is inherited as an X-linked disease. Evaluation of the patient should begin with a careful history for related family conditions or prior ocular trauma. Consideration should be given to whether the pigmentary changes are congenital or acquired. Fundus changes may be similar throughout generations in inherited disorders, and identification of these patterns may prompt medical attention. A complete medical history may reveal a systemic disorder associated with the observed fundus pigmentation, such as that seen with Gardner syndrome or with metastatic lesions to the choroid. Previous surgical intervention may alter the fundus appearance and produce diagnostic confusion without knowledge of the ocular history. Slit-lamp evaluation of the anterior segment, followed by dilated fundus examination with biomicroscopy or with a contact lens, can demonstrate changes in pigmentation with a stereoscopic advantage. Adjustment of the illumination beam may allow localization of the pigmentation within the layers of the posterior pole and show the extent of elevation. Indirect ophthalmoscopy with scleral depression can demonstrate the extent of fundus pigmentation and reveal any additional pathology. External transillumination can demonstrate the margins and the anterior extent of a pigmented lesion. Fluorescein angiography is useful in delineating the retinal circulation and demonstrating variations in the retinal pigmentation. Generally, areas of increased pigmentation are hypofluorescent throughout the phases of the angiogram. This hypofluorescence is due to static blockage of the underlying choroidal flush by the pigmentation, which may be helpful in differentiating vascular or inflammatory lesions from strictly pigmented areas. Indocyanine green (ICG) angiography is useful to demonstrate the choroidal circulation and to identify choroidal pathology. Areas of increased fundus pigmentation are typically hypofluorescent in the early phase ICG angiogram. ICG angiography is most useful for differentiating choroidal hemangioma from other choroidal lesions. The choroidal hemangioma shows hyperfluorescence during the venous phase with late hypofluorescence (washout phenomenon). Demonstration of choroidal melanoma may show an abnormal choroidal circulation and marginal late dye leakage.1 Subretinal neovascularization may also be demonstrated with ICG as a hyperfluorescent hot spot or a larger plaque. Ultrasonography is an extremely useful ancillary tool to evaluate pigmented lesions. A B-scan ultrasound can identify elevated pigmented lesions and quantitatively measure their height and basal dimensions. It can also identify subretinal fluid associated with elevated pigmented areas. Ultrasound technology, A-scan, is able to measure the axial length of an eye and further support, for example, the diagnosis of pigmentation associated with axial myopia. Internal reflectivity of an elevated pigmented lesion is discerned with the A-scan, and this may aid in distinguishing a malignant from a benign lesion. Ultrasound biomicroscopy (UBM) is a high-resolution measure of the anterior segment anatomy that uses high frequencies in the 40 to 100 Mhz range to produce images to a depth of approximately 2 to 6 mm in the anterior chamber. This tool is useful in demonstrating pathology of the trabecular meshwork and angle, ciliary body, iris, and other anterior segment structures. Another test that may be helpful in diagnosing ocular disease associated with abnormal fundus pigmentation is the electroretinogram (ERG). This test measures the electrical activity and interaction of the photoreceptors with other retinal components. It is a reflection of all rods or cones of the fundus in response to a brief flash of light. Rod and cone responses may be isolated through various testing techniques and dark or light adaptation. The ERG may be reduced or extinguished in forms of retinitis pigmentosa, and it plays a critical role in the diagnosis of this disorder. The electrooculogram (EOG) is a test to measure the electrical potential generated by the RPE cells. It is most specific for involvement of the RPE when other studies show the retina to be normal. It is most useful to confirm the diagnosis of autosomal dominant vitelliform dystrophy (Best disease). Optical coherence tomography (OCT) is an imaging technique that is analogous to ultrasound B-scan, except optical rather than acoustic reflectivity is measured. A cross-sectional image of the retina with high resolution up to 10 μ in the Z-axis is obtained with OCT. OCT is useful to assess retinal pathology such as macular thickening and to evaluate the clinical course and response to therapy. Fundus pigmentation can present in a number of forms, thus offering diagnostic challenges to the ophthalmologist. These areas can be classified by whether they are congenital or acquired, whether the vision is normal or reduced, and by the location and characteristics of the lesion. A systematic approach based on clinical observation and the use of ancillary testing can lead to the proper diagnosis and direct treatment (Table 8–1). Congenital lesions are generally stable and do not cause progressive visual loss. Pigmented lesions from birth are derived from neural crest cells and represent abnormal RPE location or aggregation. Congenital lesions may cause visual disturbances if the optic nerve or macula is involved. In rare cases, the visual loss may be due to vascular compromise or accumulation of subretinal fluid. Lesions located on the optic nerve head should be examined for distinguishing characteristics such as color, border, elevation, and appearance on ancillary testing (Fig. 8–1). A melanocytoma is often a jet-black accumulation of pigment covering or adjacent to the optic nerve head (Fig. 8–2). This is a congenital lesion with fibrillated edges, and it may be slightly elevated. The feathery edges may be due to interspersion of pigmented cells in the nerve fiber layer. It comprises magnocellular nevus cells that are darkly pigmented and differ from the slender melanosomes found in other pigmented lesions. Angiographically, the area is hypofluorescent in the early phases. Elevation noted by slit-lamp biomicroscopy can be confirmed by B-scan ultrasonography. The clinical characteristics can be found in Table 8–2. Other lesions include combined hamartoma of the retina and RPE, nevi, congenital hypertrophy of the RPE (CHRPE), and acquired hyperplasia of the RPE.
Pigmented Lesions of the Fundus: Discrete Small to Medium Size
What Is “Normal” Fundus Pigmentation?
How Are Fundus Pigmentary Changes Evaluated?
What Role Does Ancillary Testing Have in Identifying Small to Medium Discrete Lesions of the Fundus?
How Are Dark Brown or Black Pigmented Lesions of the Fundus Categorized?
What Is the Differential Diagnosis of Optic Nerve or Peripapillary Pigmented Lesions?
| Topics or Elements to Include | Reason |
|---|---|
| In the patient history: Family history Prior eye trauma or surgery Myopia | Inherited forms of RPand Gardner Syndrome Hyperplasia of retinal pigment epithelium, laser, or cryotherapy scars Lacquer cracks, Fuchs spot, lattice degeneration |
| In the examination: Slit-lamp biomicroscopy Indirect ophthalmoscopy | Level of pigmentation can guide diagnosis, determine extent of elevation of lesion Identification of peripheral lesions and evaluation for retinal breaks |
| Ancillary testing Fluorescein angiography Indocyanine green angiography B-scan ultrasonography Electroretinogram | Double circulation in choroidal melanoma Early hyperfluorescence in choroidal hemangioma Demonstrates subretinal neovascularization Hyperfluorescence during the venous phase with late hypofluorescence in choroidal hemangioma Defines lesion elevation and internal reflectivity Long axial length associated with pigmentary changes in myopia Reduced or extinguished in retinitis pigmentosa Variable in pattern dystrophies |
Does a Melanocytoma Cause Visual Symptoms?
An enlarged blind spot, often asymptomatic, may be noted on visual field testing. Infrequently, blurring of vision may occur if there is associated subretinal fluid extending to the macula. Vascular occlusions with resultant visual loss have been reported as a result of the constrictive nature of enlarging melanocytomas.2,3 No treatment is recommended unless there is growth documented by serial examinations and photographs. Although an optic nerve melanocytoma is usually benign, enlargement on occasion may signal malignant transformation. This finding has been rarely reported in the literature.4,5
How Do Combined Hamartoma of the Retina and Retinal Pigment Epithelium Present?
A similar lesion located on the optic nerve head or the peripapillary area is a combined hamartoma of the retina and RPE; Table 8–3 lists the clinical characteristics. Presenting symptoms may be strabismus, leukocoria, or a painless decrease in vision; most lesions are discovered during childhood. The pigmentation is deep gray to black and may have associated preretinal gliosis. This discrete small to medium-sized pigmented area also has adjacent vascular tortuosity and superficial telangiectatic vessels. Subretinal fluid accumulation and retinal striae may cause moderate to severe visual loss if the optic nerve or macula is involved. Generally, these lesions are minimally elevated. Fluorescein angiography demonstrates early hypofluorescence resulting from the pigmentation. Late hyperfluorescence and leakage from the abnormal vessels are observed and are characteristic of the combined hamartoma of the retina and RPE.6–8 There may be areas of retinal capillary nonperfusion peripheral to the lesion.
ARE THERE ANY SYSTEMIC DISORDERS ASSOCIATED WITH COMBINED HAMARTOMA OF THE RETINA AND RETINAL PIGMENT Epithelium?
An association has been described between persons with neurofibromatosis type II (NF2) and this ocular lesion.9,10 A less common association has been reported with NF1. When there is not an established diagnosis of NF2 and a combined hamartoma of the retina and RPE is found, imaging studies may display bilateral acoustic neuromas. There is no local potential for this lesion to undergo malignant transformation. This lesion often is noted in the posterior pole, but peripheral retinal involvement is also possible. Visual symptoms are more common when the location is peripapillary. Treatment usually consists of observation, although pars plana vitrectomy and epiretinal membrane removal have been attempted with overall poor visual recovery.
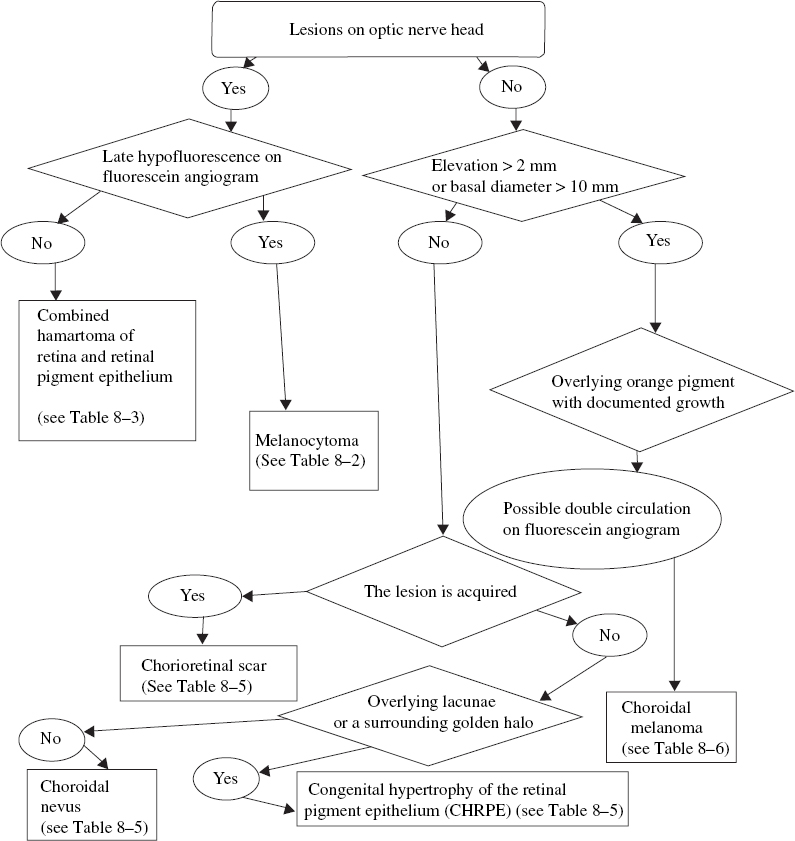
FIGURE 8–1. Clinical pathway: Small to medium-sized pigmented lesions on the optic nerve or peripapillary area. CHRPE, congenital hypertrophy of the retinal pigment epithelium.
Is There an Acquired Pigmented Lesion of the Peripapillary Area?
Peripapillary acquired pigmented lesions that are well circumscribed are also encountered, and the clinical characteristics can aid in the diagnosis. Chorioretinal scars in the posterior pole may appear as distinct areas of pigmentation. This acquired hyperplasia of the RPE is a reaction to trauma or disruption of the underlying Bruch membrane. Trauma, inflammation, or neovascularization may cause the chorioretinal scars. The lesions may expand but usually do not cause visual symptoms unless the macula is involved or recurrent subretinal neovascularization occurs. Fluorescein angiography demonstrates early hypofluorescence in the areas of pigmentation without late leakage. There is no treatment for areas of pigmentation that represent a stable scarring process.
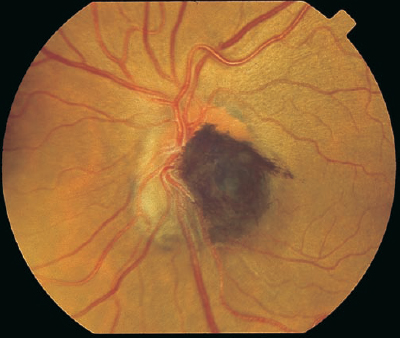
FIGURE 8–2. Jet-black pigmentation with feathered edges overlying the optic nerve characteristic of a melanocytoma.
Are There Other Peripapillary Pigmented Lesions That Are Relatively Flat?
Areas of pigmentation found on or adjacent to the optic nerve may represent a unique type of CHRPE (Table 8–4). Although more commonly found in the retinal periphery, this accumulation of pigmented cells may be found at the optic nerve head. The clinical appearance is usually that of a dark-brown to black area, frequently with overlying lacunae. CHRPE may extend slightly over the optic nerve, but it does not appear centered over the papilla. A surrounding golden halo may be present. Histopathologically, the pigment granules within the hypertrophied RPE cells are of normal size and do not demonstrate mitoses. The choriocapillaris and choroid are normal. The surrounding halo represents RPE cell depigmentation or dropout.
| Jet black to brown pigmentation |
| Flat to mildly elevated |
| Fibrillated edges |
| Minimal visual symptoms, possible enlarged blind spot |
| Hypofluorescent on fluorescein angiography |
| Stable to minimal growth over time |
| Deep gray to black pigmentation |
| Associated preretinal gliosis |
| Adjacent vascular tortuosity and telangiectatic vessels |
| Retinal striae |
| Early hypofluorescence due to pigment followed by late hyperfluorescence |
| Possible adjacent retinal vascular nonperfusion |
| Some cases associated with neurofibromatosis type II |
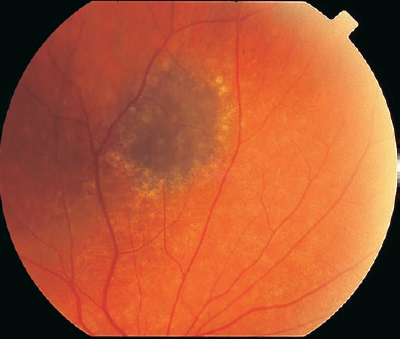
A choroidal nevus is the most common intraocular tumor observed on dilated fundus examination, and the location may be found on or adjacent to the optic nerve head (Table 8–5). A choroidal nevus is flat to minimally elevated and usually displays distinct borders (Fig. 8–3). Retinal vessels overlying the nevus may demonstrate a variable appearance from normal to sclerotic attenuation. The basal diameter is usually less than 10 mm, and there may be drusen overlying the brown to black pigmentation. Hypofluorescence in the early phase of the fluorescein angiogram is common because of the choroidal pigmentation and the lack of internal vessels. Binocular examination for elevation is key for distinguishing a potential progressive malignant process from a stable benign lesion. Documentation of a lack of growth, clinically and photographically, is also important in establishing the proper diagnosis.
DO PERIPAPILLARY CHOROIDAL NEVI CAUSE VISUAL SYMPTOMS?
Choroidal nevi can cause visual symptoms if there is associated subretinal fluid.11 The fluid extending to the macula may cause metamorphopsia and generalized visual loss. Laser demarcation along the posterior edge of the nevus, with careful attention to the optic nerve head and the macula, may be successful in walling the fluid. Laser delivered at low power to create light treatment to the nevus also may be helpful in resorption of the subretinal fluid over time. Choroidal neovascular membranes (CNV) also may arise from a choroidal nevus and produce visual symptoms.12 These CNV can be treated with laser photocoagulation. Photodynamic therapy may be considered for subfoveal CNV. Although subretinal fluid or CNV associated with choroidal nevi is uncommon, the treatment for these may pose a challenge. Choroidal nevi associated with foveal RPE derangement also cause visual symptoms. Generally, observation and monitoring of asymptomatic choroidal nevi are recommended, with use of photographs to evaluate for growth of suspicious lesions.
| Elevation less than 2 mm |
| Brown to jet-black appearance |
| Usually less than 10 mm basal diameter |
| Hypofluorescence on fluorescein angiography due to pigment |
| Overlying lacunae and surrounding halo |
| Generally well-defined borders |
| Brown to black pigmentation |
| Possible overlying drusen or orange hue |
| Elevation less than 2 mm and basal diameter usually less than 5 mm |
| Generally normal overlying vessels |
| Hypofluorescence on fluorescein angiography and indocyanine green angiography |
| Possible associated subretinal fluid with decreased vision |
What Clinical Features Increase Suspicion That a Pigmented Choroidal Lesion Is a Choroidal Melanoma?
Growth is one of the factors triggering suspicion for the development of choroidal melanoma from a preexisting choroidal nevus. Most choroidal melanomas arise de novo, with only an estimated one in 5000 originating from a choroidal nevus.13 Tables 8-6 and 8-7 review the features of lesions suspicious for choroidal melanoma and features predictive of growth.14 The lesion is suspicious when it exceeds 2 mm in height or 10 mm in basal diameter. Penetration through the Bruch membrane may allow for a “mushroom” or “collar-button” pattern of growth with dispersion of pigmented cells into the vitreous. Pigmentation may be similar to the choroidal nevus, but overlying orange pigmentation is more commonly seen in choroidal melanomas than nevi. Subretinal fluid may accumulate in association with a choroidal melanoma, producing visual loss. Overlying retinal vessels also may have a variable appearance. There is no pathognomonic pattern on fluorescein angiography for a choroidal melanoma, and the appearance depends on the amount of pigmentation, the size, and the associated subretinal fluid; however, the angiogram may help to exclude other simulating disorders. Large choroidal melanomas may display a “double circulation” pattern, with simultaneous fluorescence of the retinal and choroidal vessels in the early venous phase. Fundus photography to document the appearance of the lesion along with B-scan ultrasonography to define the dimensions should be performed. The A-scan ultrasound will demonstrate low to medium internal reflectivity in a choroidal melanoma.